Limites de l’œdème
Coordonné par C. Creuzot-Garcher
F. Devin, E. Chbat, C. Deschasse, Y. Kauffmann, M. Picco
➤ Le fovéoschisis du myope fort (FSMF) est une pathologie assez fréquente du myope fort dont l’existence n’entraîne pas nécessairement de baisse de vision.
➤ La plupart des FSMF sont découverts fortuitement lors d’un examen par tomographie par cohérence optique.
➤ L’appréciation de leur retentissement fonctionnel et la surveillance de leur évolutivité sur une durée prolongée sont indispensables.
➤ La prise en charge chirurgicale des formes sévères permet d’obtenir des résultats globalement favorables.
Le fovéoschisis du myope fort (FSMF) est une pathologie dégénérative maculaire qui conduit au développement de cavités kystiques dans les couches de la neurorétine. Il a été décrit pour la première fois en 1938 par Rochon-Duvigneaud, puis étayé en 1958 par Phillips sous la forme d’un décollement de rétine non rhegmatogène localisé au pôle postérieur, survenant volontiers en cas de staphylome myopique [1, 2]. Cependant, le FSMF est resté longtemps sous-diagnostiqué, en raison des limitations de l’examen du fond d’œil et de l’angiographie dans le cadre des myopies dégénératives. Il aura fallu attendre 1999 et l’essor de la tomographie par cohérence optique (optical coherence tomography [OCT]) pour que Takano et Kishi affinent sa définition, en le caractérisant par un clivage intrarétinien en région maculaire pouvant être associé à un décollement des photorécepteurs [3].
L’incidence du FSMF est mal connue et probablement sous-estimée en raison de l’absence de signes fonctionnels chez certains patients myopes forts, n’aboutissant pas à la réalisation systématique d’un OCT [4]. Elle est ainsi très variable selon les séries, allant de 8 à 34 % [5–7].
De nombreux éléments biomécaniques et dégénératifs contribuent à la physiopathogénie du FSMF créant progressivement étirement, dissociation des couches rétiniennes avec des phases d’aggravation plus ou moins rapides. Outre son apport pour le diagnostic des FSMF, l’OCT a contribué à une meilleure compréhension de sa physiopathologie. Le FSMF est une forme particulière des syndromes tractionnels maculaires. Dans la myopie forte, la rétine se trouve dans une situation particulièrement instable, étirée et distendue par des forces opposées. Une étude de Vanderbeek et al. en 2012 visant à identifier les différents types de tractions exercées sur la rétine du myope fort a relevé quatre mécanismes intriqués : un détachement vitréen périfovéal postérieur ; une non-conformité de la membrane limitante interne (MLI) native ; une membrane épirétinienne (MER) ; une couche vitréenne corticale résiduelle après le détachement vitréen postérieur [8]. D’autres auteurs ont évoqué l’implication d’éléments intrinsèques à la rétine amincie avec une MLI tendue et des artérioles rétiniennes raccourcies. D’une façon schématique, on identifie :
une traction tangentielle, considérée comme passive, faisant intervenir les MER, la rigidité de la MLI et les tractions exercées par les vaisseaux eux-mêmes :
les MER exercent une composante tractionnelle maculaire participant à la formation d’un FSMF. Leur fréquence est de l’ordre de 30 % en cas de FSMF, fréquence plus élevée que chez les yeux myopes sans FSMF [6],
la rigidité de la MLI serait favorisée par la prolifération de fibres de collagène et de débris cellulaires à sa surface. En OCT, cette rigidité se manifeste par un décollement de la MLI par rapport aux couches rétiniennes plus profondes [9, 10],
enfin la composante vasculaire serait exercée principalement par les artérioles rétiniennes devenues rigides, qui apparaissent sous la forme de microplis horizontaux en OCT. Ces microplis forment de petits épaississements rétiniens focaux au niveau de la rétine interne, avec un aspect de rétine tendue entre deux [6]. Cet aspect OCT persiste même après la chirurgie maculaire, les vaisseaux rétiniens étant moins à même de se réappliquer avec le reste de la rétine. Leur incidence est ainsi très variable, selon la réalisation ou non d’une vitrectomie avec pelage de la MLI, allant de 2,9 à 60 % [11, 12] ;
une traction antéropostérieure centrifuge, considérée elle aussi comme passive, constituée par le staphylome postérieur et l’allongement scléral progressif, qui a tendance à emporter la rétine externe. Cette ectasie postérieure augmente graduellement avec l’âge et l’allongement axial du globe oculaire [3, 7]. L’apparition d’un staphylome se fait en général vers 40 ans et augmente sensiblement les contraintes mécaniques sur la rétine, en induisant des changements anatomiques dans l’interface vitréomaculaire aboutissant au développement d’un schisis (fig. 15-1a) pouvant évoluer vers un trou maculaire (fig. 15-1b) ;
une traction antéropostérieure centripète, dite active, exercée par les adhérences vitréomaculaires [6, 7]. Ces tractions vitréomaculaires semblent d’autant plus fréquentes chez le myope fort que la hyaloïde postérieure est souvent très adhérente chez ces patients. La persistance de plages adhérentes de cortex vitréen postérieur est très fréquemment retrouvée en cours de vitrectomie. Ces constatations peropératoires permettent de redresser de faux diagnostics cliniques de décollement postérieur du vitré posés en préopératoire. La contraction du cortex vitréen est à l’origine d’un déplacement vers l’avant de la rétine [13, 14]. La présence d’une MER renforce l’adhésion vitréomaculaire et s’oppose au détachement spontané du vitré et de la hyaloïde postérieure, majorant la traction antérieure et aggravant la prolifération fibrocellulaire et la contraction.
Dans une étude de Bando et al., des fibres de collagène et des débris cellulaires ont été identifiés à la surface interne de la MLI exfoliée des yeux avec FSMF dans 70 % des cas [10]. Ces cellules synthétisent des fibres de collagène initiant une réponse proliférative sur la MLI. Celle-ci, rigidifiée, majore la traction statique tangentielle et antérieure s’opposant à l’adaptation rétinienne sur le staphylome postérieur et contribue au développement du FSMF et son évolution vers un trou maculaire.
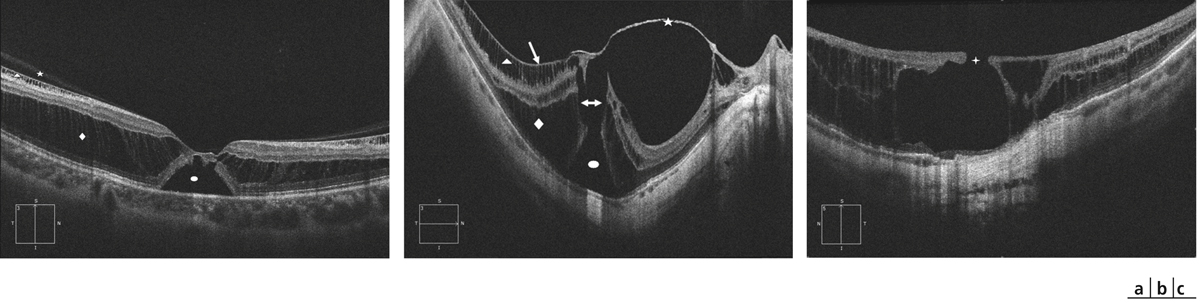
Fig. 15-1 Fovéoschisis myopiques compliqués : de décollement séreux rétinien fovéolaire (a) ; d’un DSR et d’un trou maculaire de pleine épaisseur (b) ; de trou maculaire lamellaire et atrophie choriorétinienne postérieure (c).
Étoile : hyaloïde postérieure ; flèche : membrane limitante interne ; triangle : schisis dans la couche des cellules ganglionnaires ; losange : schisis dans la rétine externe ; ovale : décollement séreux rétinien ; croix : trou maculaire lamellaire ; flèche double : trou maculaire de pleine épaisseur.
Les FSMF présentent une symptomatologie très variable. Ils peuvent être asymptomatiques (du fait de la dégénérescence myopique choriorétinienne sous-jacente) ou être à l’origine d’une baisse d’acuité visuelle et/ou de métamorphopsies. Ces signes fonctionnels ne sont de loin pas pathognomoniques du FSMF, plusieurs pathologies isolées ou associées à la myopie pouvant en être responsables : amblyopie, atrophie choriorétinienne, rupture de la membrane de Bruch ou encore néovaisseaux sous-rétiniens (fig. 15-1c) [6].
Le fovéoschisis affecte 9 à 20 % des yeux myopes avec staphylome [15]. Le FSMF touche classiquement le myope fort connu, avec une prédominance féminine. Le patient signale une baisse visuelle progressive sur plusieurs mois voire années. Les métamorphopsies sont généralement présentes et identifiées par le patient qui, cependant, éprouve des difficultés à préciser la date d’apparition des symptômes. La vision de loin est en général naturellement limitée, en raison de la myopie forte, et reste longtemps peu modifiée. La lecture sans addition excessive devient plus « fatigante » avec une perte progressive du plaisir de lire. La lecture de très près, sans correction, n’a qu’une valeur anecdotique ne permettant qu’un déchiffrage et sous-évalue l’atteinte maculaire réelle. Le motif de consultation peut également être secondaire à une complication inaugurale, comme un scotome central brutal par apparition d’un trou maculaire ou d’un décollement de rétine du pôle postérieur. L’absence d’hémorragie sous-rétinienne ou d’une pigmentation sous-rétinienne, suspecte de néovascularisation choroïdienne, est un élément négatif important bien que les deux pathologies puissent être associées. La liquéfaction et la synérèse du vitré sont fréquentes. La visualisation d’un anneau de Weiss n’affirme jamais un décollement postérieur du vitré chez le myope fort. Dans la très grande majorité des cas, la hyaloïde postérieure, ou la hyaloïde postérieure schisique, est adhérente au pôle postérieur chez le myope fort. Curieusement dans cette pathologie dégénérative maculaire du myope fort, les lésions rhegmatogènes périphériques sont assez rares malgré la myopie forte.
Le diagnostic de FSMF reste difficile à porter par simple examen du fond d’œil. En effet, une vision nette et claire de l’interface vitréorétinienne est généralement impossible en raison de la déformation secondaire au staphylome, d’un trouble des milieux ou encore de l’absence d’uniformité de l’épithélium pigmentaire maculaire sous-jacent. Seuls les stades avancés sont facilement décelables au fond d’œil, lorsque la neurorétine est décollée de manière étendue, ou quand un trou maculaire associé ou non à un décollement de rétine postérieur s’est déjà développé [6].
À l’instar de la biomicroscopie, les rétinophotographies, l’angiographie et l’échographie en mode B ne sont que peu contributives au diagnostic positif de forme modérée de FSMF [6].
Comme l’avait démontré Gallemore, l’OCT est l’outil le plus sensible pour porter le diagnostic de FSMF. Il permet de visualiser les dissociations caractéristiques des couches rétiniennes et d’écarter certains diagnostics différentiels de baisse d’acuité visuelle dans un contexte de myopies dégénératives [16].
En OCT, le FSMF atteint l’ensemble des couches de la rétine. Il se présente classiquement sous la forme d’une rétine épaissie en région maculaire, secondaire à un clivage entre la rétine externe fine, flexible et plutôt hyporéflective, et la rétine interne plus épaisse, rigide et hyper-réflective. Au sein de cette rétine schisique, il existe un espace hyporéflectif, reliant la rétine interne à la rétine externe, associé à un aspect de colonnes tissulaires correspondant vraisemblablement aux cellules de Müller résiduelles (fig. 15-2). D’autres signes peuvent être visibles tels que des logettes d’œdème intrarétinien ou des trous lamellaires (fig. 15-3) [7, 17].
Plusieurs stades évolutifs ont été décrits. Initialement, le FSMF se présente comme un rétinoschisis pur au sein des couches nucléaires et plexiformes externes. Le retentissement fonctionnel est souvent modeste. Des acuités visuelles de l’ordre de 0,3 à 0,6 sont habituelles en l’absence d’autres anomalies maculaires. Par la suite, lorsque les forces tractionnelles exercées sur la rétine l’empêchent de se conformer à la configuration du staphylome, un décollement du neuro-épithélium fovéolaire peut apparaître. C’est à ce stade qu’une baisse d’acuité visuelle franche peut être perçue par le patient. Dans les stades plus avancés, un trou maculaire par amincissement extrême des couches externes de la fovéa, voire un décollement de rétine peuvent survenir [17].
Parmi les images OCT souvent spectaculaires, certaines sont fréquentes dans le FSMF : les images de stratifications multiples prérétiniennes correspondent à plusieurs entités histologiques avec des schisis de la MLI, des MER plus ou moins clivées et des schisis de la hyaloïde postérieure. Dans les FSMF, la rétine externe peut paraître longtemps respectée expliquant la discordance entre une fonction visuelle relativement conservée et des déstructurations sévères de la rétine interne. Certaines altérations discrètes de la rétine externe sont à rechercher en spectral-domain optical coherence tomography (SD-OCT). Le coton ball sign (fig. 15-4) décrit par Tsunoda et al. se définit par une région ronde ou diffuse hyper-réflective au centre de la fovéa entre la ligne des segments internes et latéral (portion ellipsoïde) et les terminaisons des photorécepteurs [18].
Au stade précoce du FSMF, une petite quantité de liquide sous-rétinien peut être détectée en SD-OCT, plus ou moins associée à une rupture de la rétine neurosensorielle et à une MER en plus de la traction vitréomaculaire [19, 20]. Il est ainsi possible de suivre les changements architecturaux dans les couches rétiniennes au cours de la progression de la myopie axiale et de l’étirement du globe. En SD-OCT, la vacuolisation intrarétinienne est retrouvée dans 49,5 % des yeux myopes forts, alors que leur incidence en ophtalmoscopie n’est que de 24,4 % [21]. Le swept-source OCT permettrait de mieux voir les structures rétinochoroïdiennes, le vitréoschisis et le schisis périphérique. La morphologie du FSMF peut être classée en deux types : avec ou sans détachement fovéolaire, selon les forces d’adhésion cellulaire, plus ou moins affaiblies entre les cellules de la rétine neurosensorielle et le niveau d’atrophie de la rétine.
À partir de ces éléments, il convient de distinguer deux présentations différentes de FSMF avec des implications thérapeutiques différentes :
le FSMF isolé, sans décollement de la couche des photorécepteurs ou trou maculaire. Le retentissement fonctionnel est souvent modeste, le patient conserve son acuité de lecture sans addition excessive. Des acuités visuelles de 20/30 à 20/60 sont habituelles en l’absence d’autres atteintes liées à la myopie forte. L’association d’un staphylome postérieur conditionne l’évolutivité. Son absence est un facteur de stabilité qui peut être très prolongé, tandis que sa présence et sa sévérité sont deux éléments de mauvais pronostic qui oriente vers une aggravation prévisible ;
le FSMF compliqué soit d’un décollement de la couche des photorécepteurs, soit d’une ouverture isolée de la couche des photorécepteurs (trou maculaire de la rétine externe) ou d’un trou maculaire de pleine épaisseur, soit par association de ces formes. L’apparition d’un décollement de la couche des photorécepteurs plus fréquemment retrouvé dans les yeux avec staphylome postérieur et/ou atrophie choriorétinienne (fig. 15-5) est très souvent symptomatique s’accompagnant d’une baisse visuelle perçue par le patient. En cas de vision centrale déjà détériorée, il peut toutefois être asymptomatique. Ce décollement constituerait un facteur de risque non négligeable d’apparition d’un trou maculaire secondaire (fig. 15-1b).
Dans la plupart des cas ne seront considérées que les baisses d’acuité visuelle rendant la lecture difficile voire impossible avec une addition conforme à l’âge. Les indications opératoires, tant que cette acuité autorise un vrai Parinaud 4, sont exceptionnelles. La situation de l’œil adelphe est importante mais une vision très basse de celui-ci ne doit pas faire écarter une option chirurgicale utile. L’association d’un FSMF à un décollement séreux rétrofovéolaire de la couche des photorécepteurs doit inciter à considérer une chirurgie avant que n’apparaisse un trou maculaire de pleine épaisseur. La présence d’un trou maculaire compliquant un FSMF justifie dans la plupart des cas une chirurgie.
Une biométrie ultrasonique en mode A ou B permet de conforter la sévérité de la myopie axiale en particulier chez le pseudophaque. Des longueurs axiales supérieures à 33 mm doivent être documentées avant une éventuelle indication chirurgicale, afin que l’instrumentation adaptée soit disponible (longueur de pince suffisante).
Si l’angiographie à la fluorescéine n’a pas d’intérêt dans le diagnostic positif du FSMF, elle peut être utile dans les cas litigieux. Dans certains cas de suspicion de néovaisseaux choroïdiens rétrofovéolaires du myope fort, en particulier s’il existe une pigmentation sous-rétinienne ou des pétéchies hémorragiques postérieures, cet examen précisera l’activité néovasculaire. L’association FSMF et néovaisseaux choroïdiens actifs modifie l’approche thérapeutique et complète l’information éclairée du patient et de l’ophtalmologiste référent.
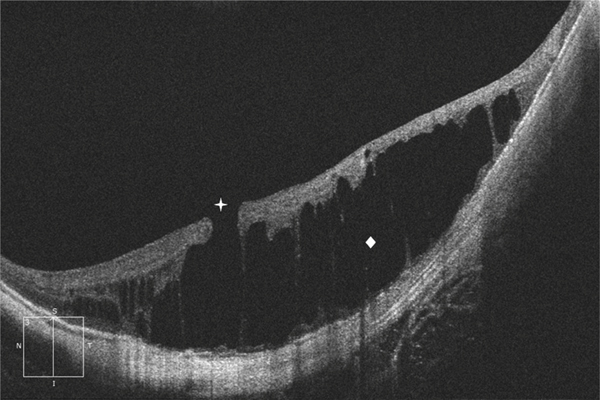
Fig. 15-2 Fovéoschisis myopique sans décollement fovéolaire avec un rétinoschisis externe et trou maculaire lamellaire.
Losange : schisis dans la rétine externe ; croix : trou maculaire lamellaire
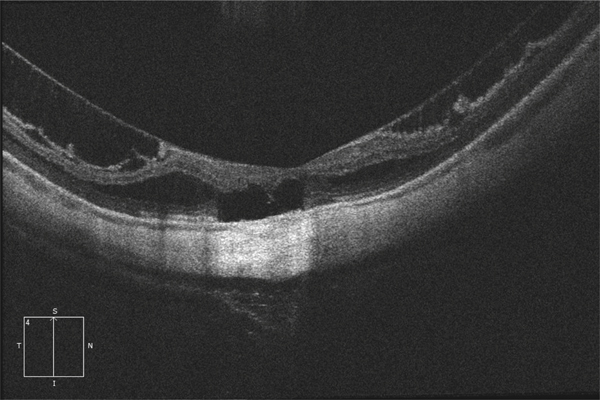
Fig. 15-3 Fovéoschisis myopique compliqué d’une ouverture de la rétine externe (trou maculaire externe).
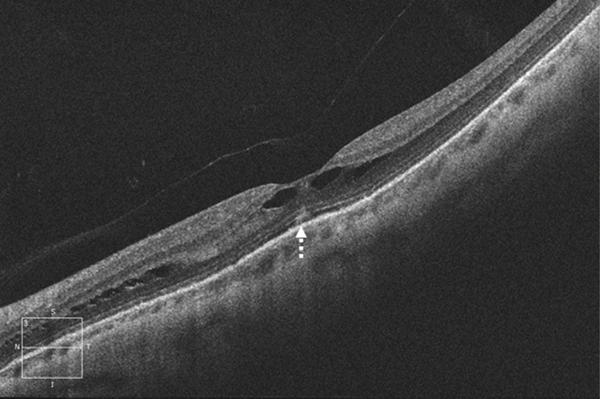
Fig. 15-4 Fovéoschisis myopique rétinien non compliqué avec aspect de coton ball en OCT.
Flèche en pointillé : trou maculaire de la rétine externe.
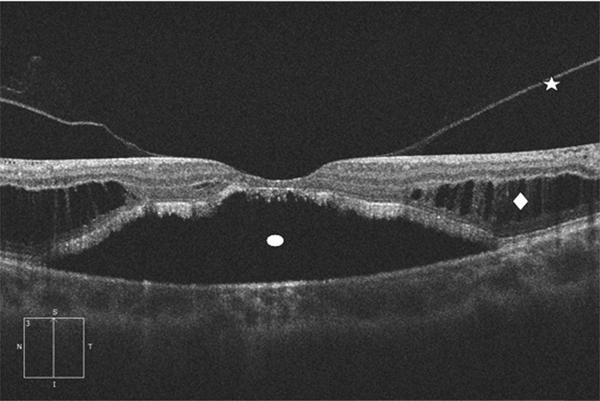
Fig. 15-5 Fovéoschisis myopique compliqué d’un DSR étendu avec atrophie choriorétinienne.
Étoile : hyaloïde postérieure ; losange : schisis dans la rétine externe ; ovale : décollement séreux rétinien.
Le traitement de choix du FSMF est la vitrectomie permettant de lever toutes les contraintes tractionnelles exercées sur la macula, afin qu’elle puisse retrouver une morphologie moins altérée en épousant la forme du staphylome. Il n’y a pas actuellement de consensus sur les indications chirurgicales. Le traitement du FSMF n’est jamais une urgence (sauf en cas de décollement rétinien étendu), la rétine d’un myope fort semblant paradoxalement capable de supporter des contraintes qui seraient intolérables chez les non-myopes. En l’absence d’autres complications maculaires, il est licite d’en poser l’indication chirurgicale lorsqu’il existe une aggravation fonctionnelle documentée sur plusieurs mois ou une aggravation anatomique majeure (augmentation de la hauteur du FSMF ; apparition d’un décollement fovéolaire, d’un trou maculaire ou d’un décollement de rétine) [17, 22, 23].
Dans un premier temps opératoire, un décollement complet et soigneux de la hyaloïde postérieure doit être réalisé. Les résidus du cortex vitréen peuvent être identifiés à l’aide de l’acétonide de triamcinolone et retirés en exerçant des mouvements de traction tangentielle. Dans un second temps, toute MER présente est disséquée [11]. Le pelage de la MLI reste actuellement sujet à controverse et délicate en raison de la mauvaise visibilité et des conditions opératoires. Spaide et Fisher estiment qu’il augmenterait le risque de développer un trou maculaire en postopératoire pour ces yeux ayant déjà une fovéa des plus fines [14]. D’autres auteurs en proposent le pelage en dedans des arcades vasculaires sur deux à trois diamètres papillaires, soit en première intention, soit après échec d’une première chirurgie, en faisant valoir son rôle non négligeable dans la physiopathologie du FSMF. Ce geste est actuellement facilité par l’usage de colorants vitaux tel le bleu de Coomassie (Brilliant Blue G) [11, 12]. Plus récemment, des auteurs japonais ont proposé un pelage de la limitante épargnant la fovéa sans jamais atteindre le bord de celle-ci. Cette technique limite la traction exercée par la MLI et ainsi le risque d’arrachage de la rétine lors du passage en pont au-dessus du toit fragile de la rétine schisique [24]. Pour certains, un tamponnement par gaz peut être effectué associé au maintien du patient en position « bulle » pendant la semaine suivant l’intervention [11, 23]. Son indication est discutée au cas par cas. Indiqué en cas de trou maculaire idiopathique associé, le tamponnement n’est pas indispensable pour permettre une réapplication d’un FSMF associé ou non à un décollement séreux rétrofovéolaire et/ou une rupture de la rétine externe. Pour certains, il augmenterait même l’incidence de trou maculaire secondaire [25].
Plusieurs complications postopératoires peuvent survenir, comme l’accélération rapide d’une cataracte, le développement d’une atrophie choriorétinienne ou l’apparition d’un trou maculaire ou d’un décollement de rétine [17, 22, 23].
Le standard idéal doit permettre de ménager un compromis entre l’efficacité de la vitrectomie centrale (simplifiée par la liquéfaction du vitré) et les contraintes d’une dissection postérieure rigoureuse sur des longueurs axiales augmentées tout en minimisant l’effraction sclérale. Certains instruments « rigidifiés » peuvent poser des problèmes de longueur pour accéder au pôle postérieur. La disponibilité d’instruments rallongés est parfois indispensable surtout concernant les pinces. Dans le cas de trou maculaire de grand diamètre (> 400 µ), il a été proposé d’utiliser un lambeau de MLI pour fermer mécaniquement le trou maculaire. Avec cette technique, les pourcentages de clôture s’élèvent à 98 % par rapport à 88 % en pelage de MLI traditionnel [26]. Le drainage par aspiration du liquide sous-rétinien au niveau d’un éventuel trou maculaire est à proscrire sauf dans le cas d’un décollement de rétine étendu. Si une intervention combinée à une phaco-exérèse est réalisée, la prescription d’un myotique en postopératoire limitera les risques de synéchies iridocapsulaires et le patient sera positionné en évitant le décubitus dorsal jusqu’à résorption complète du gaz. En effet, la zonule est souvent distendue chez le myope fort avec un risque élevé de passage de gaz en chambre antérieure. En cas de trou maculaire associé, le positionnement sera plus strict et prolongé que dans les trous maculaires idiopathiques (de l’ordre de 70 à 80 % du temps durant 7 à 12 jours). Les taux de fermeture des trous maculaires associés au FSMF sont variables (de 25 à 87 % selon les études), le succès semblant dépendre avant tout de la profondeur du staphylome [21].
La vitrectomie et les dissections prémaculaires nécessitent de parfaites conditions de visualisation. Dans cette pathologie, l’indication d’une chirurgie combinée phaco-exérèse/vitrectomie systématique se discute. En cas d’opacification nucléaire du cristallin dont le retentissement sur la visualisation peropératoire est très fréquemment sous-estimé, il faudra systématiquement l’associer. Elle pose toutefois la problématique de l’anisométropie chez un patient souvent assez jeune et phaque sur son œil adelphe.
L’indentation maculaire en traitement de première intention du FSMF a été proposée par Baba en 2006 [27]. Elle inverse la courbure du staphylome et permet de traiter les décollements fovéolaires associés au FSMF. Plusieurs techniques sont utilisables avec soit la mise en place d’une indentation localisée (éponge, cale de Ando, etc.), soit selon un principe d’indentation antéropostérieure par bande supportant une cale maculaire type T macular buckle de Devin-Morin [28, 29]. Cette technique permet le positionnement d’une indentation postérieure sans avoir les contraintes d’une exposition directe (pas de dépose du muscle droit latéral nécessaire) et sans avoir à placer des sutures postérieures sur une sclère amincie (fig. 15-6).
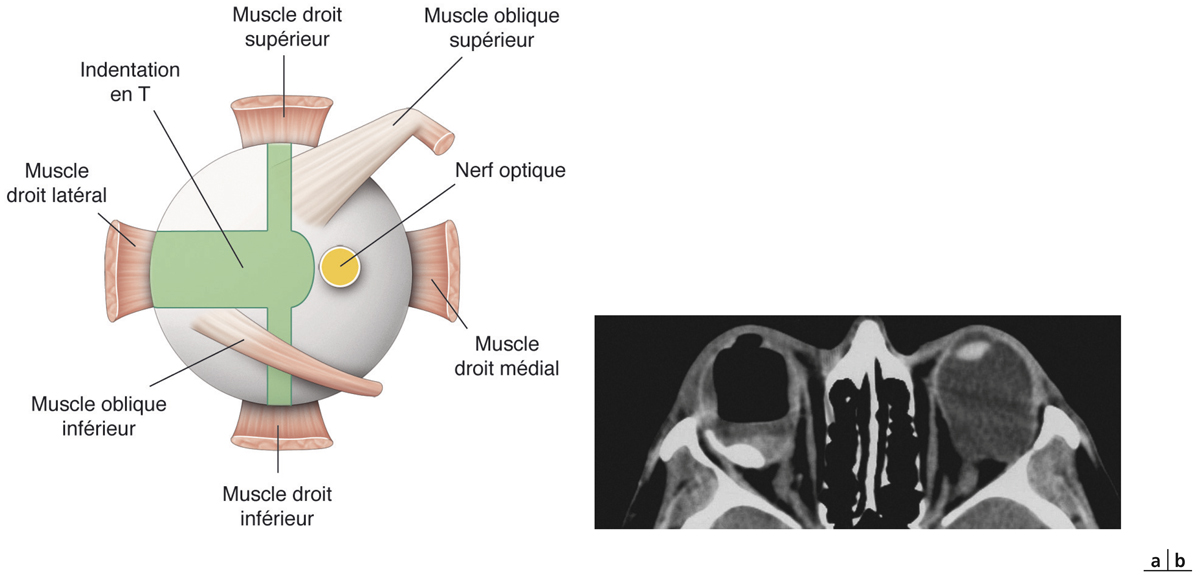
Fig. 15-6 Positionnement de l’indentation en T.
a. Positionnement de l’indentation maculaire en T par rapport à l’insertion des muscles oculomoteurs (rose) et au nerf optique (jaune). b. OD : contrôle du bon positionnement de l’indentation en T (en vert) par scanner (ou écho B). OD : aphaque, initialement sous silicone ; celui-ci a pu être retiré en même temps que la mise en place de l’indentation avec un tamponnement provisoire par air. OG : noter la déformation ectasique du pôle postérieur et l’abrasion mécanique sur l’orbite créée par les mouvements oculaires.
L’évolution naturelle du FSMF est difficilement prédictible. Cependant, la plupart des séries ont retrouvé un déclin fonctionnel, avec une baisse d’acuité visuelle dans 69 % des cas et l’apparition de métamorphopsies dans 55 % des cas au terme de 2 ans de suivi en l’absence de traitement [17]. Près de la moitié de ces patients évolueraient vers un trou maculaire ou un décollement de rétine après 3 ans de suivi [30]. Deux facteurs de progression ont été identifiés : la présence d’une traction maculaire (par adhérence vitréomaculaire ou par MER) et la présence d’un décollement du neuro-épithélium fovéolaire [17].
La chirurgie a pour objectif de s’opposer à ce déclin fonctionnel. Elle permettrait, dans le meilleur des cas, à un gain de plus de 2 lignes de Snellen dans 55 % des cas et à une régression du FSMF à l’OCT dans 73 % des cas [17]. Les résultats chirurgicaux sont souvent retardés et la récupération peut se poursuivre sur une période de 6 mois à plus de 1 an (fig. 15-7 à 15-9). Les facteurs de bon pronostic sont l’acuité visuelle préopératoire conservée, une courte durée d’évolution de la symptomatologie, et une longueur axiale inférieure à 28 mm [22, 31]. Les facteurs de mauvais pronostic seraient une épaisseur du FSMF supérieure à 500 µm, la présence d’une traction vitréomaculaire et/ou d’un décollement fovéolaire et l’existence d’un trou maculaire et/ou d’un décollement de rétine [17].
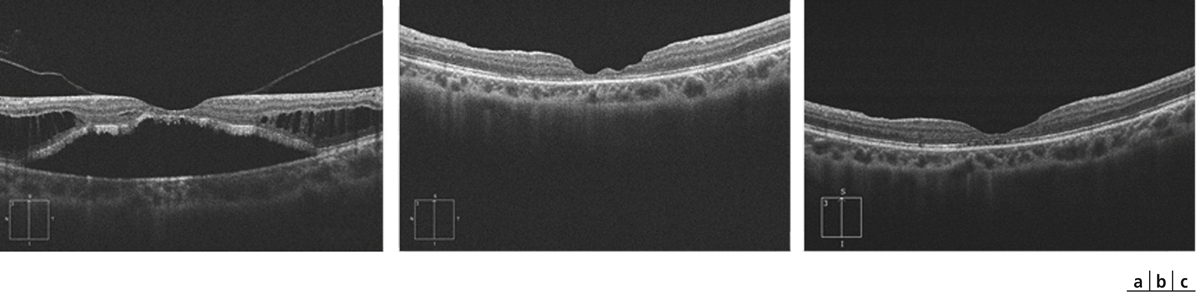
Fig. 15-7 Fovéoschisis du myope fort avec décollement séreux.
Aspect préopératoire (a), réapplication rétinienne après vitrectomie sans tamponnement par gaz : aspects postopératoires à 2 et 6 mois (b, c). L’acuité visuelle s’améliore de 20/63 p5 à 20/40 p2.
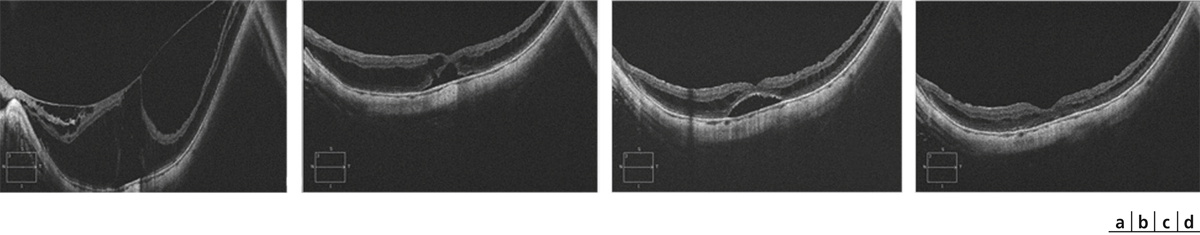
Fig. 15-8 Fovéoschisis du myope fort.
Aspect préopératoire (a), réapplication après vitrectomie sans tamponnement gazeux – à 13 jours (b), 3 mois (c) et 16 mois postopératoires (d). L’acuité visuelle s’améliore de 20/100 p8 à 20/32 p3 avec légère métamorphopsie à 17 mois postopératoire.
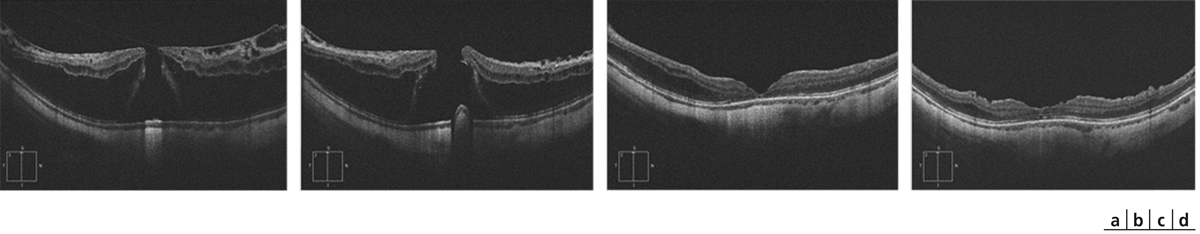
Fig. 15-9 Fovéoschisis du myope fort compliqué de trou maculaire et de décollement rétinien.
Aspect préopératoire (a, b), réapplication après vitrectomie + tamponnement par gaz : aspect postopératoire à 1 mois et 1 an (c, d). L’acuité visuelle s’améliore de 20/200 p14 à 20/63 p6.
[1] Rochon-Duvigneaud M. Déformation et lésions de l’oeil myope. Bulletin de la Société d’Ophtalmologie de Paris 1938 ; 1-10.
[2] Phillips CI. Retinal detachment at the posterior pole. Br J Ophthalmol 1958 ; 42 : 749-53.
[3] Takano M, Kishi S. Foveal retinoschisis and retinal detachment in severely myopic eyes with posterior staphyloma. Am J Ophthalmol 1999 ; 128 : 472-6.
[4] Ichibe M, Baba E, Funaki S, et al. Retinoschisis in a highly myopic eye without vision impairment. Retina 2004 ; 24 : 331-3.
[5] Baba T, Ohno-Matsui K, Futagami S, et al. Prevalence and characteristics of foveal retinal detachment without macular hole in high myopia. Am J Ophthalmol 2003 ; 135 : 338-42.
[6] Panozzo G, Mercanti A. Optical coherence tomography findings in myopic traction maculopathy. Arch Ophthalmol 2004 ; 122 : 1455-60.
[7] Benhamou N, Massin P, Haouchine B, et al. Macular retinoschisis in highly myopic eyes. Am J Ophthalmol 2002 ; 133 : 794-800.
[8] VanderBeek BL, Johnson MW. The diversity of traction mechanisms in myopic traction maculopathy. Am J Ophthalmol 2012 ; 153 : 93-102.
[9] Sayanagi K, Ikuno Y, Tano Y. Tractional internal limiting membrane detachment in highly myopic eyes. Am J Ophthalmol 2006 ; 142 : 850-2.
[10] Bando H, Ikuno Y, Choi JS, et al. Ultrastructure of internal limiting membrane in myopic foveoschisis. Am J Ophthalmol 2005 ; 139 : 197-9.
[11] Ikuno Y, Gomi F, Tano Y. Potent retinal arteriolar traction as a possible cause of myopic foveoschisis. Am J Ophthalmol 2005 ; 139 : 462-7.
[12] Sayanagi K, Ikuno Y, Gomi F, Tano Y. Retinal vascular microfolds in highly myopic eyes. Am J Ophthalmol 2005 ; 139 : 658-63.
[13] Kobayashi H, Kishi S. Vitreous surgery for highly myopic eyes with foveal detachment and retinoschisis. Ophthalmology 2003 ; 110 : 1702-7.
[14] Spaide RF, Fisher Y. Removal of adherent cortical vitreous plaques without removing the internal limiting membrane in the repair of macular detachments in highly myopic eyes. Retina 2005 ; 25 : 290-5.
[15] Tang J, Rivers MB, Moshfeghi AA, et al. Pathology of macular foveoschisis associated with degenerative myopia. J Ophthalmol 2010 ; 2010, pii : 175613.
[16] Gallemore RP, Jumper JM, McCuen BW 2nd, et al. Diagnosis of vitreoretinal adhesions in macular disease with optical coherence tomography. Retina 2000 ; 20 : 115-20.
[17] Gaucher D, Haouchine B, Tadayoni R, et al. Long-term follow-up of high myopic foveoschisis : natural course and surgical outcome. Am J Ophthalmol 2007 ; 143 : 455-62.
[18] Tsunoda K, Watanabe K, Akiyama K, et al. Highly reflective foveal region in optical coherence tomography in eyes with vitreomacular traction or epiretinal membrane. Ophthalmology 2012 ; 119 : 581-7.
[19] Sergeev YV, Caruso RC, Meltzer MR, et al. Molecular modeling of retinoschisin with functional analysis of pathogenic mutations from human X-linked retinoschisis. Hum Mol Genet 2010 ; 19 : 1302-13.
[20] Lim LS, Cheung G, Lee SY. Comparison of spectral domain and swept-source optical coherence tomography in pathological myopia. Eye (Lond) 2014 ; 28 : 488-91.
[21] Bures-Jelstrup A, Alkabes M, Gomez-Resa M, et al. Visual and anatomical outcome after macular buckling for macular hole with associated foveoschisis in highly myopic eyes. Br J Ophthalmol 2014 ; 98 : 104-9.
[22] Kumagai K, Furukawa M, Ogino N, Larson E. Factors correlated with postoperative visual acuity after vitrectomy and internal limiting membrane peeling for myopic foveoschisis. Retina 2010 ; 30 : 874-80.
[23] Kwok AK, Lai TY, Yip WW. Vitrectomy and gas tamponade without internal limiting membrane peeling for myopic foveoschisis. Br J Ophthalmol 2005 ; 89 : 1180-3.
[24] Shimada N, Sugamoto Y, Ogawa M, et al. Fovea-sparing internal limiting membrane peeling for myopic traction maculopathy. Am J Ophthalmol 2012 ; 154 : 693-701.
[25] Kim KS, Lee SB, Lee WK. Vitrectomy and internal limiting membrane peeling with and without gas tamponade for myopic foveoschisis. Am J Ophthalmol 2012 ; 153 : 320-326.e1.
[26] Garcia-Layana A, Garcia-Arumi J, Ruiz-Moreno JM, et al. A review of current management of vitreomacular traction and macular hole. J Ophthalmol 2015 ; 2015 : 809640.
[27] Baba T, Tanaka S, Maesawa A, et al. Scleral buckling with macular plombe for eyes with myopic macular retinoschisis and retinal detachment without macular hole. Am J Ophthalmol 2006 ; 142 : 483-7.
[28] Ando F, Ohba N, Touura K, Hirose H. Anatomical and visual outcomes after episcleral macular buckling compared with those after pars plana vitrectomy for retinal detachment caused by macular hole in highly myopic eyes. Retina 2007 ; 27 : 37-44.
[29] Devin F, Tsui I, Morin B, et al. T-shaped scleral buckle for macular detachments in high myopes. Retina 2011 ; 31 : 177-80.
[30] Shimada N, Ohno-Matsui K, Baba T, et al. Natural course of macular retinoschisis in highly myopic eyes without macular hole or retinal detachment. Am J Ophthalmol 2006 ; 142 : 497-500.
[31] Ikuno Y, Sayanagi K, Soga K, et al. Foveal anatomical status and surgical results in vitrectomy for myopic foveoschisis. Jpn J Ophthalmol 2008 ; 52 : 269-76.
V. Caillaux, D. Gaucher
➤ La macula bombée (MB) est une protrusion convexe de la macula présente généralement au sein du staphylome postérieur d’un œil myope fort.
➤ La hauteur du dôme progresse avec l’évolution du staphylome myopique et de l’atrophie sclérochoroïdienne.
➤ On note un épaississement scléral en regard du bombement et un décollement séreux rétinien fluctuant.
➤ L’OCT réalisé en coupes multidirectionnelles permet de définir trois grandes morphologies de macula bombée.
➤ Aucun traitement n’a à ce jour fait la preuve de son efficacité dans les baisses de vision liée à la MB.
La macula bombée (dome-shaped macula) ou MB est une anomalie anatomique du pôle postérieur du myope fort, décrite pour la première fois en 2008 par Gaucher et al. [1]. Elle se complique fréquemment d’un décollement sous-rétinien. L’origine de ce décollement sous-rétinien est mal connue, mais il correspond à un épaississement rétinien et pourrait résulter d’un trouble de la barrière hémato-rétinienne. À ce titre, la MB peut entrer dans la liste des causes d’œdème maculaire ou pour le moins en constituer l’un des diagnostics différentiels. Le décollement sous-rétinien de la macula bombée est d’ailleurs fréquemment confondu avec des néovaisseaux choroïdiens du myope fort. Savoir reconnaître cette entité permet de ne pas traiter à tort des patients par des injections intravitréennes inutiles.
La MB est définie par une protrusion convexe, en dôme, de la macula au sein du staphylome postérieur d’un œil myope fort. Cette entité n’est pas rare, elle affecte environ 10 % des yeux myopes forts [1, 2].
Différentes hypothèses ont été émises pour expliquer le développement d’une MB : résistance localisée à la déformation du staphylome scléral, épaississement choroïdien focal dans la région maculaire, hypotonie oculaire, invagination sclérale au niveau d’un effondrement de la partie postérieure de la paroi oculaire ou encore traction vitréorétinienne tangentielle [1, 3]. Finalement, les analyses en enhanced depth imaging–optical coherence tomography (EDI-OCT) ont mis en évidence un épaississement scléral relatif et focal, au centre de la macula, en regard du bombement, sans déformation de courbure de la paroi externe du globe oculaire [2, 4]. Une étude récente a montré que le bombement progressait dans le temps [5]. L’augmentation du bombement serait en fait due à la progression du staphylome myopique et de l’atrophie des tissus sclérochoroïdiens autour de la région maculaire. Le « creusement » du staphylome autour d’une macula indemne expliquerait l’augmentation de la hauteur de bombement [5, 6].
La survenue d’un décollement sous-rétinien au sommet du bombement maculaire est une complication relativement fréquente des MB. Sa prévalence est très variable selon les séries : de l’ordre de 10 % dans les séries asiatiques [2, 4] à près de 50 % dans les séries européennes [1, 6, 7]. Ces variations importantes pourraient être liées aux caractéristiques des populations étudiées : origines ethniques différentes, recrutement en centres plus ou moins spécialisés. La physiopathogénie du décollement sous-rétinien n’est pas encore élucidée. Il pourrait s’expliquer par une perturbation de l’écoulement des fluides à partir de la choroïde secondaire à l’épaississement scléral focal. L’épaississement scléral excessif conduirait à un remaniement compressif de la choroïde, lui-même responsable d’un dysfonctionnement de l’épithélium pigmentaire de la rétine. Il en résulterait la formation d’un décollement sous-rétinien à partir de points de fuite [4, 7, 8]. L’hypothèse d’un épaississement choroïdien focal associé à une hyperperméabilité choroïdienne, comparable à ce qui est observé dans la choriorétinopathie séreuse centrale (CRSC), est aussi avancée [1, 7].
On observe une nette prédominance féminine dans toutes les séries publiées. L’atteinte est bilatérale dans la moitié des cas [1, 6]. La réfraction moyenne des patients atteints de MB se situe autour de −10 dioptries avec des extrêmes allant de −1,50 à −23 dioptries [1, 2, 6]. Il n’est ainsi pas impossible de constater une MB dans des yeux présentant une faible myopie. En outre, une étude récente a évoqué le diagnostic dans des yeux emmétropes voire hypermétropes [9]. Il n’a pas été mis en évidence de corrélation entre l’importance de la longueur axiale ou de l’erreur réfractive et la présence d’une MB [2].
Dans la majorité des cas, le diagnostic est posé chez un patient consultant pour une baisse d’acuité visuelle ou des métamorphopsies. La macula bombée peut néanmoins être asymptomatique et découverte fortuitement [1].
L’acuité visuelle au moment du diagnostic est modérément altérée, habituellement supérieure à 4/10 [2, 10]. La présence d’un décollement sous-rétinien est associée à une moins bonne acuité visuelle [6].
L’examen du fond d’œil est rarement évocateur du diagnostic et est rendu difficile par les altérations du fond d’œil associées à la myopie forte (choroïdose myopique, conus, plages d’atrophie choriorétinienne, ruptures de la membrane de Bruch). Il peut mettre en évidence des migrations pigmentaires maculaires (fig. 15-10) et parfois une anomalie de courbure de la macula lorsque la protrusion est importante. La protrusion maculaire se constitue au sein du staphylome myopique. Le DSR, habituellement fin, est difficile à visualiser au sein des remaniements maculaires.
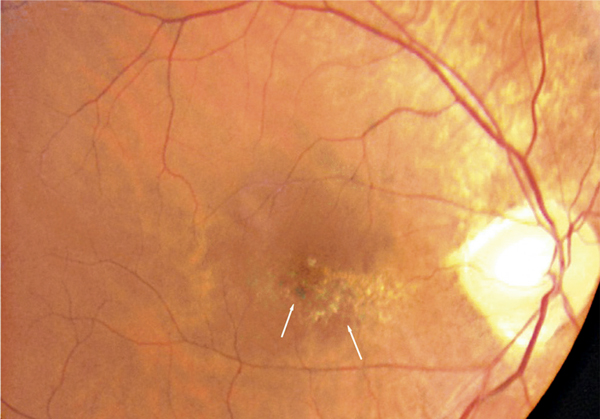
Fig. 15-10 Rétinographie en couleurs d’une macula bombée typique.
La macula est le siège de remaniements pigmentés et atrophiques (flèches).
La tomographie en cohérence optique (optical coherence tomography [OCT]) est l’examen clé pour le diagnostic de MB. Il met en évidence une protrusion interne, en dôme, au sein du staphylome, au niveau maculaire. Cette protrusion est secondaire à une saillie de la rétine neurosensorielle, de l’épithélium pigmentaire et de la choroïde centrée sur la fovéa. La courbure concave de la face interne du pôle postérieur devient convexe au niveau du bombement maculaire. Il est important de réaliser des coupes OCT multidirectionnelles, aussi bien dans l’axe horizontal que dans l’axe vertical, le bombement n’étant parfois visible que sur un seul axe. Les reconstructions en trois dimensions sont particulièrement intéressantes pour visualiser le bombement et analyser sa morphologie au sein du staphylome.
Trois types morphologiques de MB ont été décrits selon l’orientation de l’axe principal du bombement au sein du staphylome (fig. 15-11) [6] :
le dôme circulaire correspond à une protrusion en calotte sphérique, centrée par la fovéa. La macula est clairement convexe sur les coupes OCT verticales et horizontales. Le dôme est entouré par le staphylome dans toutes les directions. Il s’agit de la forme la plus typique, mais ne représentant que 20 % des cas ;
le bombement ovale horizontal correspond à une protrusion ovalaire à grand axe horizontal, séparant le staphylome myopique en deux parties, supérieure et inférieure. La macula est convexe sur les coupes OCT verticales, alors qu’elle apparaît « plate » sur les coupes OCT horizontales. Il s’agit du type le plus fréquemment observé (60 à 80 % des cas) ;
le bombement ovale vertical correspond à une protrusion ovalaire à grand axe vertical, séparant le staphylome myopique en deux parties, nasale et temporale. La macula est convexe sur les coupes OCT horizontales, alors qu’elle apparaît « plate » sur les coupes OCT verticales. Il s’agit du type le moins fréquent. Il se compliquerait plus fréquemment de décollement sous-rétinien [6].
L’OCT permet également de mettre en évidence un décollement sous-rétinien (fig. 15-12), localisé au sommet de la protrusion. Plus rarement, le décollement sous-rétinien peut être déclive et réaliser en angiographie un aspect de coulée gravitationnelle, prenant alors un aspect évocateur de CRSC chronique. Ce décollement sous-rétinien est habituellement peu important en épaisseur. La couche des photorécepteurs située au contact du décollement sous-rétinien prend un aspect irrégulier d’autant plus que le décollement sous-rétinien est ancien. Le décollement sous-rétinien est parfois associé à un fin décollement de l’épithélium pigmentaire non fibrovasculaire [2, 7]. Il existe une corrélation positive entre la hauteur du bombement maculaire et la présence d’un décollement sous-rétinien. De plus, l’épaisseur choroïdienne est d’autant plus importante que la hauteur du bombement est importante [6]. L’épaisseur choroïdienne semble plus importante dans la région fovéolaire, au niveau du bombement, qu’en bordure du staphylome. Néanmoins, le lien entre épaisseur choroïdienne et décollement sous-rétinien demeure incertain : l’étude de Viola et al. [7] a mis en évidence une épaisseur choroïdienne fovéolaire augmentée dans les yeux avec décollement sous-rétinien, ce que n’a pas retrouvé l’étude de Caillaux et al. [6].
Les nouveaux appareils d’OCT (EDI et swept-source) permettent une bonne visualisation de la sclère chez les myopes forts. Il a ainsi été mis en évidence un épaississement scléral localisé en regard du bombement maculaire [4]. L’importance de l’épaississement scléral n’est pas corrélée à la survenue d’un décollement sous-rétinien [2].
Le diagnostic de décollement sous-rétinien compliquant une MB peut s’avérer difficile et nécessiter la réalisation d’autres examens d’imagerie maculaire. Il doit être distingué des autres causes de décollements sous-rétiniens maculaires, tels que la néovascularisation choroïdienne, la CRSC, la vasculopathie polypoïdale choroïdienne ou les tumeurs choroïdiennes.
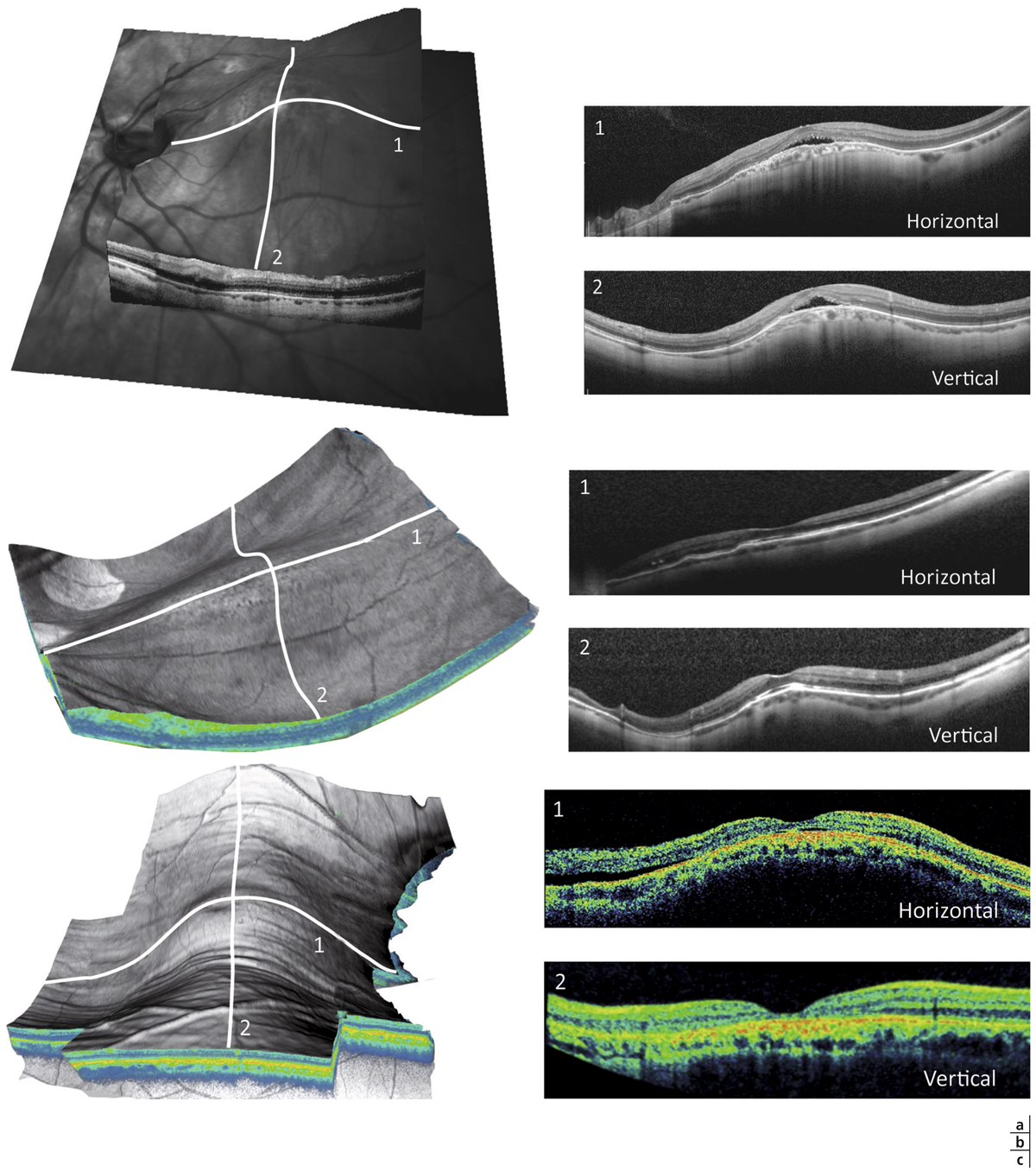
Fig. 15-11 Aspects OCT et formes tridimensionnelles des différentes maculas bombées.
On distingue la forme en dôme circulaire (a). Les coupes verticale (a1) et horizontale (a2) sont typiques et montrent une protrusion maculaire, avec une inversion localisée, convexe et régulière de la courbure du globe touchant la sclère, la choroïde et la rétine. La forme ovale horizontale (b) correspond à une protrusion ovalaire d’axe horizontal, séparant le staphylome myopique en deux parties, supérieure et inférieure ; seule la coupe OCT verticale (b : 2) est typique. La forme ovale verticale (c) sépare le staphylome en deux verticalement ; seule la coupe OCT horizontale (c : 1) est typique.
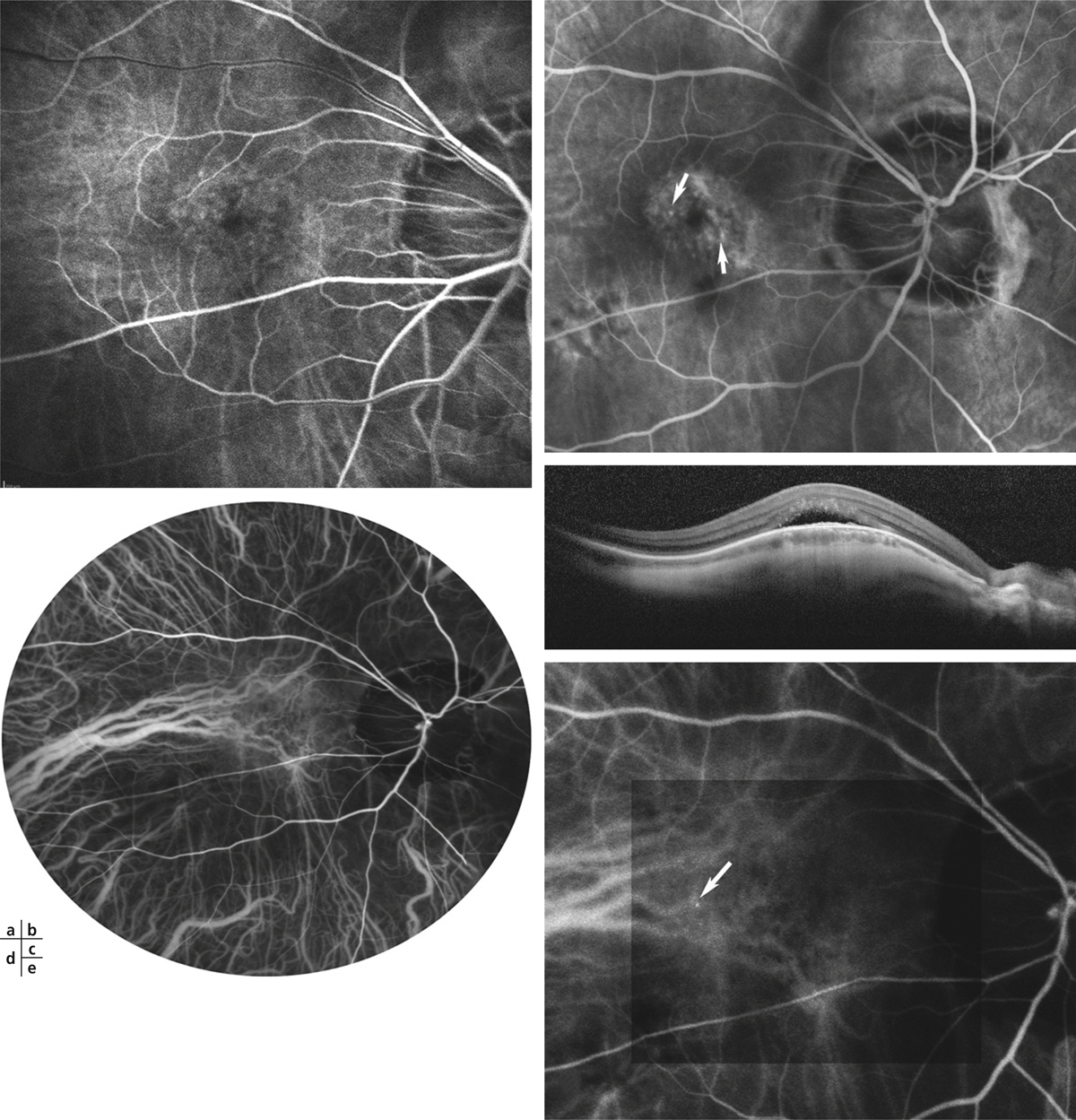
Fig. 15-12 Aspects angiographiques de la macula bombée.
En angiographie à la fluorescéine (FA), on note une hyperfluorescence précoce due à un effet fenêtre au niveau de l’épithélium pigmentaire atrophique (a). Les points de fuite en tête d’épingle (pin points) sont très fréquents au temps tardif de la FA (b, flèches) en cas de décollement sous-rétinien (c). L’angiographie au vert d’indocyanine (ICG) est souvent normale, elle ne montre pas d’hyperperméabilité des vaisseaux choroïdiens (d). Au temps tardif de l’ICG, on voit parfois des points de fuite en tête d’épingle (e, flèche) qui sont aussi liés à la présence d’un décollement sous-rétinien.
En angiographie à la fluorescéine (fluorescein angiography [FA]), la macula est le siège d’une fluorescence hétérogène, liée à l’association d’altérations atrophiques de l’épithélium pigmentaire, hyperfluorescentes par effet fenêtre, et de migrations pigmentaires hypofluorescentes par effet masque. Le remplissage d’un décollement sous-rétinien est souvent difficile à mettre en évidence en FA, des points de fuite de type pin points sont parfois visibles (fig. 15-12). En angiographie au vert d’indocyanine (indocyanine green [ICG]), les altérations de l’épithélium pigmentaire et de la choriocapillaire maculaire se traduisent par une hypofluorescence tardive.
Les points de fuite hyperfluorescents en FA sont parfois aussi visibles sous la forme d’une hyperfluorescence ponctiforme sur les temps tardifs de l’ICG, ce qui fait évoquer une atteinte choroïdienne focale dans cette pathologie (fig. 15-12). La présence d’un décollement sous-rétinien n’est pas associée à une hyperperméabilité choroïdienne étendue en angiographie ICG, mais elle est associée à ces points de fuite en FA et en ICG qui sont retrouvés dans presque 90 % des cas de décollement sous-rétinien [7].
Le bombement maculaire convexe est confirmé par l’échographie oculaire en mode B ou par l’imagerie par résonance magnétique (IRM ; fig. 15-13). Ces examens prennent tout leur intérêt en cas de doute diagnostique avec une pathologie tumorale choroïdienne.
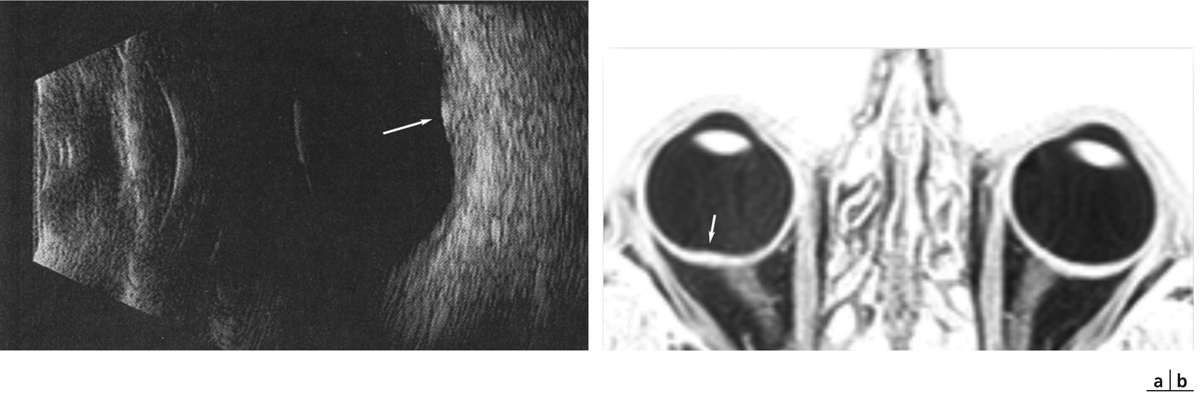
Fig. 15-13 Macula bombée en échographie et en IRM.
En échographie B, la protrusion maculaire est bien visible (a, flèche) ; en revanche, la sclère semble normale. En IRM, la protrusion est visible ainsi que l’épaississement scléral en regard de la macula (b, flèche)
Ellabban et al. [5] ont étudié l’évolution morphologique d’une série de 35 yeux atteints de MB sur une période de 2 ans. Ils ont mis en évidence une diminution significative de l’épaisseur sclérale maculaire. Cet amincissement était plus prononcé dans les secteurs périfovéolaires qu’au niveau rétrofovéolaire, à l’origine d’une augmentation de la hauteur du bombement maculaire. L’épaisseur choroïdienne a également diminué de façon significative pendant la durée du suivi. L’évolution morphologique à plus long terme des MB est encore mal connue de même que son évolution fonctionnelle.
Une seule étude a analysé le profil évolutif du décollement sous-rétinien associé aux MB [6]. Toutefois le suivi est court et sur une petite série (13 yeux suivis de 12 à 24 mois). Dans cette étude, le décollement sous-rétinien s’était résolu spontanément et durablement dans 23,1 % des cas. Dans 76,9 % des cas, son importance (épaisseur et étendue) fluctuait au cours du suivi : s’aggravant, s’améliorant, ou encore disparaissant puis réapparaissant. Ces variations n’étaient pas associées à des variations de l’épaisseur choroïdienne au niveau du bombement maculaire. L’aggravation d’un décollement sous-rétinien était associée à l’apparition de nouveaux points de diffusion en angiographie à la fluorescéine et de nouveaux points hyperfluorescents aux temps tardifs de l’angiographie ICG.
Dans les évolutions chroniques et prolongées, le décollement sous-rétinien s’accompagne d’altérations de l’épithélium pigmentaire et parfois d’un amincissement de la rétine neurosensorielle en regard.
Les hypothèses concernant la physiopathologie du décollement sous-rétinien tendent à cibler la choroïde. À l’heure actuelle, il n’y a pas de traitement validé du décollement sous-rétinien associé aux MB. La littérature est pauvre sur le sujet. Les quelques résultats portant sur des cas isolés ou de petites séries sont décevants. La photocoagulation au laser des points de diffusion angiographiques [1] et les injections intravitréennes d’anti-vascular endothelial growth factor (anti-VEGF) ne semblent pas efficaces [11, 12]. Les résultats de la photothérapie dynamique (photodynamic therapy [PDT]) sur la macula sont discutés. Une étude sur deux patients a montré une efficacité anatomique (disparition du décollement sous-rétinien) et fonctionnelle (amélioration de l’acuité visuelle) après une séance de traitement par PDT demi-fluence. Un des patients a présenté une récidive du décollement sous-rétinien 2 ans plus tard, sans réponse à une seconde PDT [12]. Récemment, une étude de deux cas a montré une résolution du décollement sous-rétinien après un traitement par anti-aldostérone (spironolactone) par voie orale [11].
Ces cas isolés ou issus de petites séries, non contrôlés, ne permettent pas de conclure quant à l’efficacité des traitements proposés et l’attitude à tenir face à un décollement sous-rétinien compliquant une MB doit rester prudente. Dans la mesure où la résolution spontanée du décollement sous-rétinien est possible dans cette pathologie, il est judicieux de ne proposer un traitement qu’aux décollements sous-rétiniens persistants ou récidivants, fréquemment et rapidement, s’ils s’accompagnent d’un retentissement sur la fonction visuelle.
[1] Gaucher D, Erginay A, Lecleire-Collet A, et al. Dome-shaped macula in eyes with myopic posterior staphyloma. Am J Ophthalmol 2008 ; 145 : 909-14.
[2] Ohsugi H, Ikuno Y, Oshima K, et al. Morphologic characteristics of macular complications of a dome-shaped macula determined by swept-source optical coherence tomography. Am J Ophthalmol 2014 ; 158 : 162-70.
[3] Mehdizadeh M, Nowroozzadeh MH. Dome-shaped macula in eyes with myopic posterior staphyloma. Am J Ophthalmol 2008 ; 146 : 478 ; author reply 478-9.
[4] Imamura Y, Iida T, Maruko I, et al. Enhanced depth imaging optical coherence tomography of the sclera in dome-shaped macula. Am J Ophthalmol 2011 ; 151 : 297-302.
[5] Ellabban AA, Tsujikawa A, Muraoka Y, et al. Dome-shaped macular configuration : longitudinal changes in the sclera and choroid by swept-source optical coherence tomography over two years. Am J Ophthalmol 2014 ; 158 : 1062-70.
[6] Caillaux V, Gaucher D, Gualino V, et al. Morphologic characterization of dome-shaped macula in myopic eyes with serous macular detachment. Am J Ophthalmol 2013 ; 156 : 958-67.
[7] Viola F, Dell’Arti L, Benatti E, et al. Choroidal findings in dome-shaped macula in highly myopic eyes : a longitudinal study. Am J Ophthalmol 2015 ; 159 : 44-52.
[8] Byeon SH, Chu YK. Dome-shaped macula. Am J Ophthalmol 2011 ; 151 : 1101 ; author reply 1101-2.
[9] Errera MH, Michaelides M, Keane PA, et al. The extended clinical phenotype of dome-shaped macula. Graefes Arch Clin Exp Ophthalmol 2014 ; 252 : 499-508.
[10] Ellabban AA, Tsujikawa A, Matsumoto A, et al. Three-dimensional tomographic features of dome-shaped macula by swept-source optical coherence tomography. Am J Ophthalmol 2013 ; 155 : 320-8.
[11] Dirani A, Matet A, Beydoun T, et al. Resolution of foveal detachment in dome-shaped macula after treatment by spironolactone : report of two cases and mini-review of the literature. Clin Ophthalmol Auckl NZ 2014 ; 8 : 999-1002.
[12] Chinskey ND, Johnson MW. Treatment of subretinal fluid associated with dome-shaped macula. Ophthalmic Surg Lasers Imaging Retina 2013 ; 44 : 593-5.
F. Metge-Galatoire
➤ Le rétinoschisis juvénile lié à l’X est la plus fréquente des dégénérescences maculaires juvéniles chez le garçon et s’accompagne d’un œdème maculaire kystique souvent associé à un rétinoschisis périphérique.
➤ L’OCT et l’électrorétinogramme (ERG) sont essentiels au diagnostic.
➤ Il existe une très grande hétérogénéité clinique y compris dans une même famille pour une même mutation.
➤ Les hémorragies intravitréennes et les décollements de rétine constituent les principales complications qui s’observent essentiellement chez le jeune enfant.
➤ L’évolution de la maladie est lente et le plus souvent compatible avec une acuité visuelle longtemps préservée.
➤ L’apparition d’une atrophie maculaire très invalidante survient en général après l’âge de 60 ans.
➤ La thérapie génique constitue un probable traitement d’avenir.
Le rétinoschisis juvénile lié à l’X est une maladie rétinienne congénitale bilatérale. Elle se caractérise par la présence d’un clivage anormal de la rétine centrale et périphérique, responsable d’une baisse visuelle progressive et de complications vitréorétiniennes à type d’hémorragie intravitréenne et de décollement de rétine. Cette affection, décrite pour la première fois par Haas en 1898 [1], s’accompagne d’un aspect maculaire assez proche de celui d’un œdème maculaire cystoïde (OMC), aussi bien cliniquement qu’en imagerie OCT. Cependant, le terrain sur lequel elle survient (l’enfant), le contexte familial pathologique et l’absence de signes angiographiques la différencient clairement de l’OMC.
Le rétinoschisis juvénile lié à l’X est la cause la plus fréquente de dégénérescence maculaire juvénile chez le garçon. Sa prévalence est estimée de façon assez variable selon la localisation géographique entre 1/5 000 à 1/25 000 [2]. Son mode de transmission est récessif lié à l’X, avec une pénétrance complète et une expressivité variable.
Le diagnostic de la maladie est souvent posé chez le garçon d’âge scolaire ou préscolaire, typiquement entre 5 et 10 ans, devant des difficultés à la lecture. C’est plus rarement un strabisme qui révèle l’affection dans la petite enfance. L’atteinte fonctionnelle est bilatérale mais le plus souvent assez nettement asymétrique [3]. Une forte hypermétropie axile est souvent associée au rétinoschisis juvénile. Il existe une très grande hétérogénéité clinique d’un individu à l’autre, mais également au sein d’une même famille, aussi bien sur le plan anatomique que sur le plan fonctionnel avec un retentissement fonctionnel plus ou moins précoce et plus ou moins important pour une même mutation.
L’acuité visuelle se détériore habituellement de façon significative pendant les deux premières décennies pour se situer autour de 2 à 5/10e, puis reste le plus souvent relativement stable jusqu’à la 5e ou 6e décennie. L’apparition secondaire d’une atrophie maculaire lentement évolutive peut s’accompagner d’une nouvelle aggravation progressive de la baisse visuelle (≤ 1/10) au-delà de 60 ans [2].
Sur le plan anatomique, le rétinoschisis juvénile lié à l’X se caractérise typiquement par un aspect d’œdème maculaire microkystique stellaire bilatéral, centré sur la fovéola, avec un aspect en « rayons de roue » ou en « pétales de fleur » qui est retrouvé dans 95 à 100 % des cas (fig. 15-14) [4]. Cet aspect a pu être décrit chez le très jeune enfant (< 3 mois) [5]. Cependant il peut être absent à un stade précoce et apparaître au cours de l’évolution de la maladie pour concerner, tôt ou tard, 100 % des patients. L’aspect typique de la macula se modifie avec le temps et prend des aspects multiples, les cavités kystiques ayant tendance à progressivement coalescer ; après 50 ans, des remaniements pigmentaires associés à un certain degré d’atrophie maculaire sont fréquents ; l’évolution ultime se fait vers l’atrophie maculaire plus ou moins étendue. L’atteinte anatomique – comme l’atteinte fonctionnelle – peut être très asymétrique chez un même individu [6].
Un rétinoschisis périphérique, unique ou multiple, le plus souvent localisé en temporal inférieur, est associé à l’atteinte maculaire dans 40 à 50 % des cas [2]. Il est initialement bulleux et transparent. Son évolution peut se faire, avant l’âge de 10 ans le plus souvent, vers la résolution spontanée, laissant place à des plages de pigmentations ou de dépigmentations périphériques, ou vers l’apparition de déhiscences dans le feuillet interne du rétinoschisis, parfois très étendues et périvasculaires, laissant certains vaisseaux dépourvus de support rétinien et flottant dans la cavité vitréenne (fig. 15-15).
La survenue d’une hémorragie intravitréenne est possible au cours de l’évolution et peut être le mode de révélation de la maladie, au point que toute hémorragie intravitréenne unilatérale chez le garçon occasionnée par un traumatisme minime ou a fortiori spontanée doit faire évoquer le diagnostic. Le décollement de rétine (DR), plus rare, est exceptionnellement inaugural.
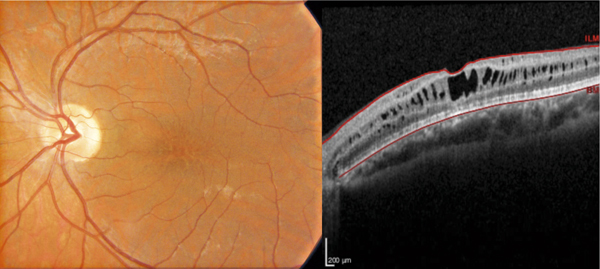
Fig. 15-14 Aspect maculaire en rayons de roue.
En OCT, clivage intrarétinien prédominant au niveau de la plexiforme interne et étendu au-delà de la zone centromaculaire.
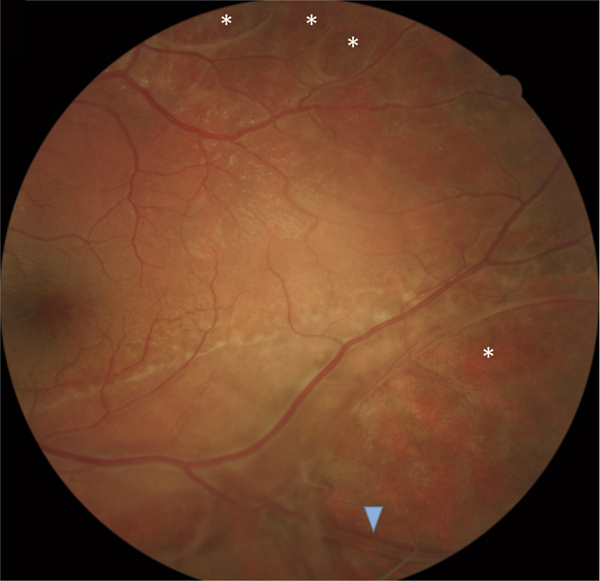
Fig. 15-15 Présence d’un rétinoschisis périphérique chez un enfant de 12 ans.
Les astérisques (*) montrent la présence de déhiscences dans le feuillet interne de la rétine. Un vaisseau flottant est visible au sein de la vaste déhiscence inférieure (flèche).
Le rétinoschisis périphérique est pourvoyeur des deux principales complications du rétinoschisis juvénile, survenant souvent lors des deux premières décennies, l’hémorragie intravitréenne et le DR (fig. 15-16) :
l’hémorragie intravitréenne et/ou intrakystique observée dans 4 à 40 % des cas est due à la rupture d’un vaisseau en pont ou plus rarement à une néovascularisation secondaire. Lorsque l’hémorragie est inaugurale, c’est l’OCT du deuxième œil qui orientera le diagnostic en montrant les anomalies maculaires caractéristiques du rétinoschisis ;
le DR complique le rétinoschisis juvénile dans 5 à 22 % des cas ; il est lié à la présence concomitante de déhiscences dans les feuillets interne et externe de la rétine. Les déhiscences du feuillet externe sont beaucoup plus rares que dans le feuillet interne, ce qui explique que le DR soit relativement peu fréquent dans le rétinoschisis juvénile [2].
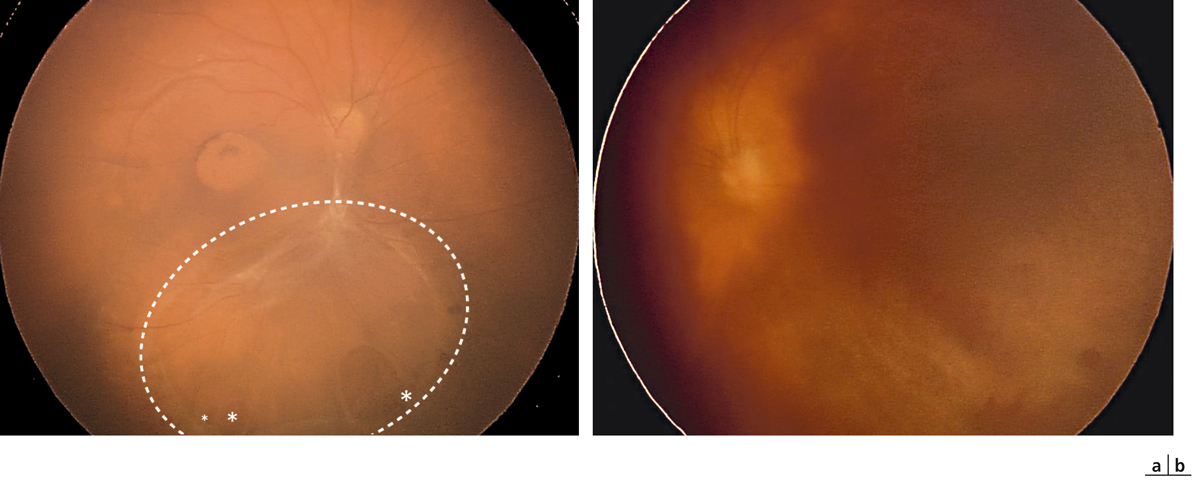
Fig. 15-16 Enfant de 14 ans présentant un rétinoschisis visible au niveau de l’œil droit (a) et une hémorragie intravitréenne de l’œil gauche (b).
La macula paraît cliniquement partiellement atrophique. On note un rétinoschisis inférieur avec de petites déhiscences dans le feuillet interne (*) et une vaste déhiscence dans le feuillet externe (indiquée par des pointillés).
Les clichés en autofluorescence montrent très clairement l’aspect microkystique radiaire en rayons de roue et isofluorescent de la macula.
L’OCT est aujourd’hui l’examen de premier plan pour le diagnostic de la maladie. En mode « en face », l’aspect en rayons de roue est particulièrement démonstratif (fig. 15-17). En coupes, l’OCT objective le clivage intrarétinien et permet de mieux analyser l’évolution de la maculopathie au cours du temps. L’image caractéristique est celle d’un clivage intrarétinien d’importance et de localisation variables (fig. 15-14, 15-18 et 15-19).
Dans une analyse détaillée récente de l’atteinte maculaire en OCT, portant sur 20 yeux de patients atteints de rétinoschisis juvénile, âgés en moyenne 17,6 ans et comparés à un groupe témoin, Yang et al. retrouvent la présence d’un fovéoschisis dans 85 % des cas et une atteinte périphérique dans 55 % des cas [7] ; 15 % des patients présentent une atteinte périphérique isolée et 45 % une atteinte fovéolaire isolée. La présence de défects dans la couche des photorécepteurs est observée dans 75 % des cas. Le rétinoschisis intéresse dans 85 % des cas les rétines interne et externe, et ce de façon égale. Au niveau de la rétine externe, les structures préférentiellement intéressées par le rétinoschisis sont la plexiforme externe (60 % des cas) et la ligne correspondant à l’extrémité des articles externes des cônes (cone outer segment tip line [COST line]) qui apparaît irrégulière dans 75 % des cas et est associée à un raccourcissement des articles externes des photorécepteurs.
La mauvaise acuité visuelle paraît nettement corrélée à l’importance des anomalies de la rétine externe (défects dans la COST line et raccourcissement des segments externes des photorécepteurs).
Au cours du temps, la maculopathie peut évoluer vers la constitution d’un kyste maculaire unique (fig. 15-20) par coalescence progressive des cavités schisiques ; des aspects de trou maculaire ou de trou lamellaire ont également pu être observés [8]. Enfin, aux stades tardifs de la maladie, apparaissent des plages d’atrophie avec disparition du clivage rétinien et amincissement de la rétine (fig. 15-21).
L’OCT est également utile pour différencier rétinoschisis et DR ou faire le diagnostic de DR associé au rétinoschisis périphérique, en particulier lorsque le rétinoschisis s’étend de façon assez postérieure. L’analyse de la rétine périphérique reste plus difficile en OCT (fig. 15-22).
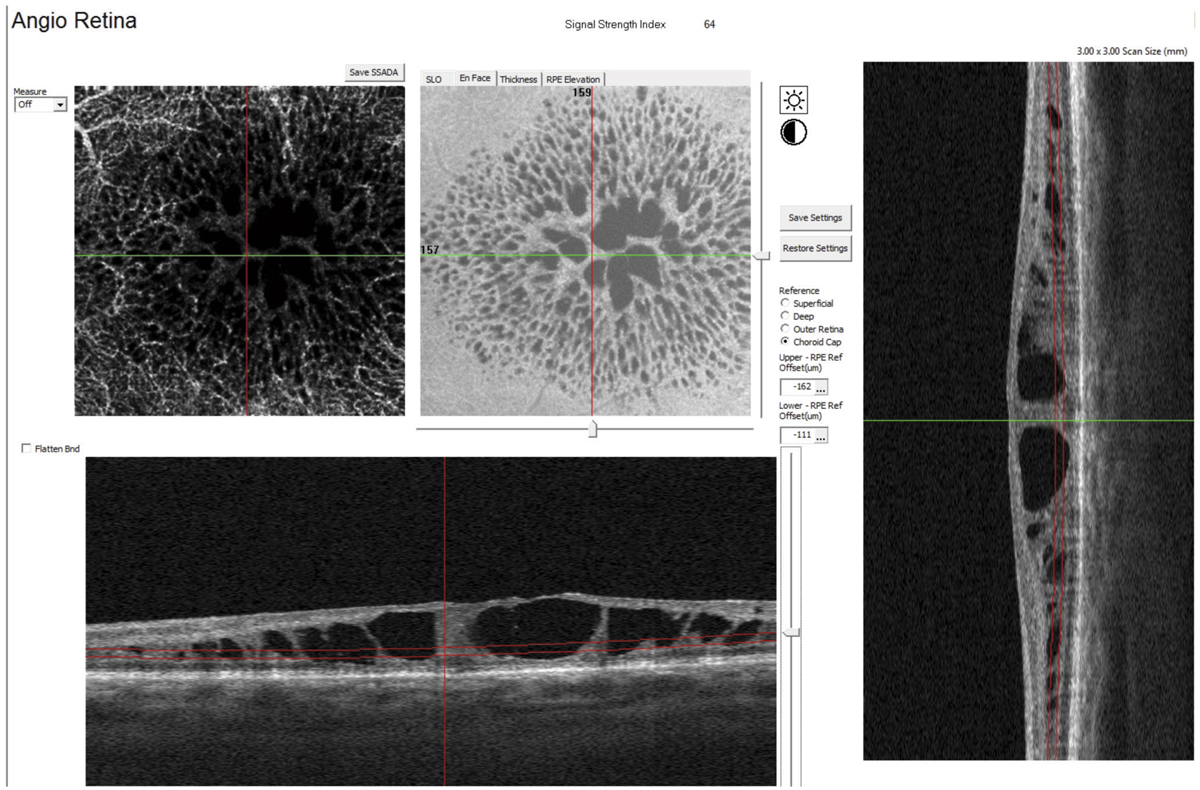
Fig. 15-17 En OCT « en face », noter l’aspect en rayons de roue et la correspondance en OCT bidimensionnel.
Cet aspect considéré hors contexte pourrait faire évoquer le diagnostic d’œdème maculaire cystoïde, principal diagnostic différentiel.
(Source : Dr B. Haouchine.)
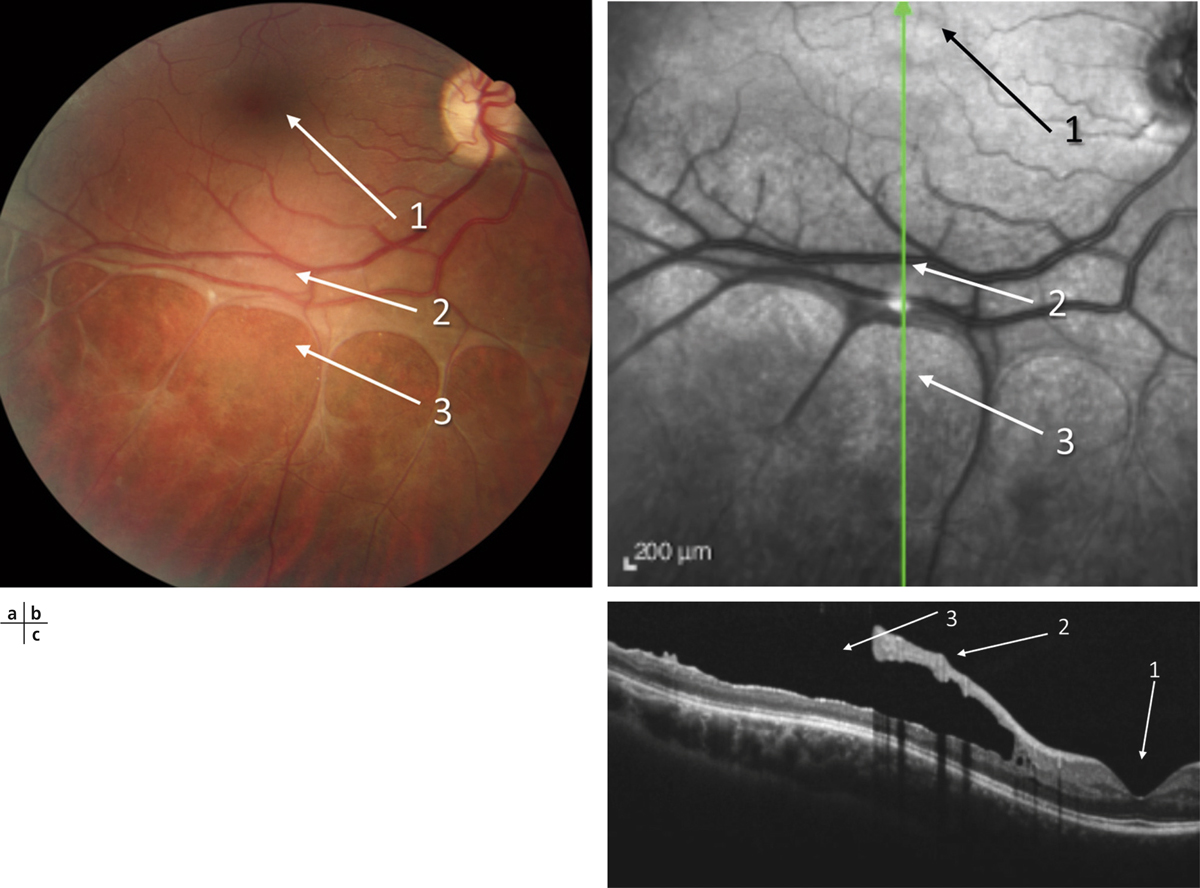
Fig. 15-18 Rétinoschisis juvénile chez un jeune homme de 21 ans dont l’acuité visuelle est de 10/10.
a. Cliché couleur. b. Cliché anérythre. c. Coupe verticale OCT.
Noter l’aspect maculaire normal (1), le rétinoschisis périphérique (2) et les déhiscences dans le feuillet interne de la rétine (3).
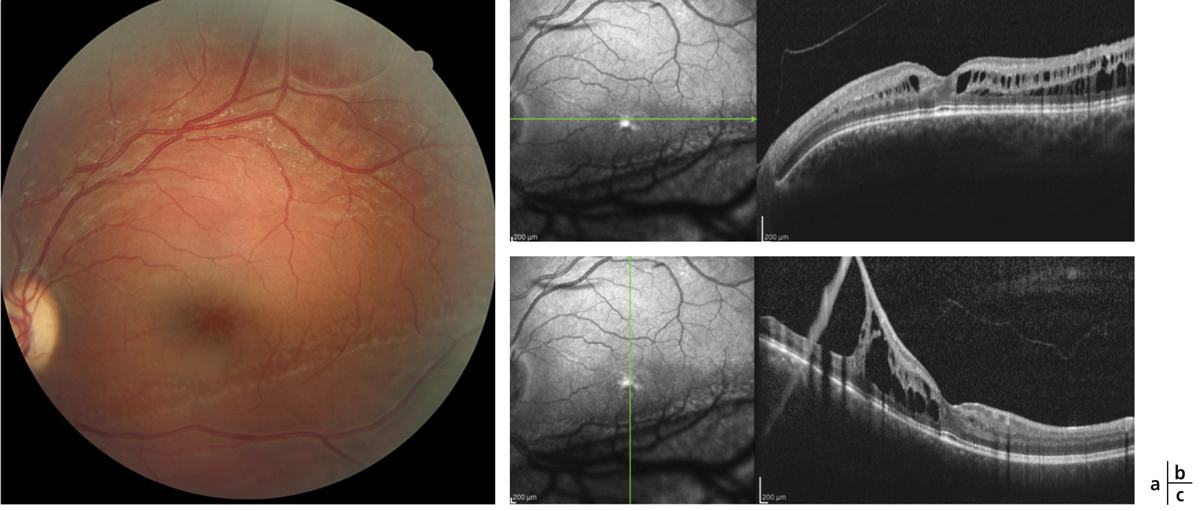
Fig. 15-19 Rétinoschisis juvénile chez un jeune patient avec acuité visuelle à 8/10.
a. Aspect maculaire caractéristique en pétales de fleur sur le cliché en couleurs. b. En coupe OCT horizontale, la dépression fovéolaire est conservée. c. Mais en coupe verticale le rétinoschisis périphérique s’étend jusqu’à la partie inférieure de la macula.
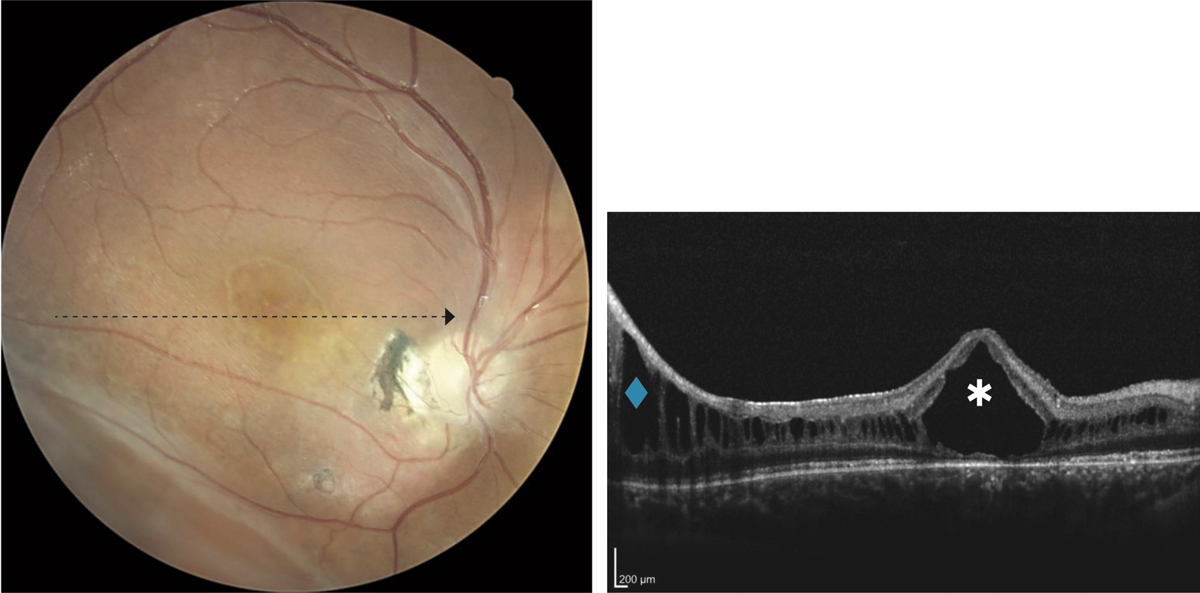
Fig. 15-20 Vaste kyste centromaculaire (*) chez un enfant de 11 ans présentant une acuité visuelle de 1/10 avec atrophie maculaire en arrière du kyste et présence d’un rétinoschisis périphérique (◆).
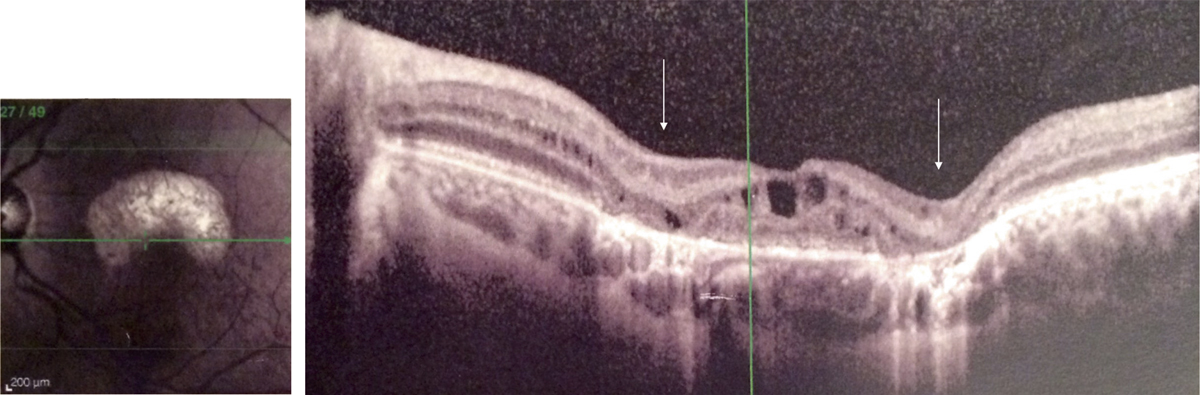
Fig. 15-21 Atrophie supéromaculaire (flèche) épargnant le centre de la macula chez un jeune homme de 17 ans présentant une acuité visuelle 5/10.
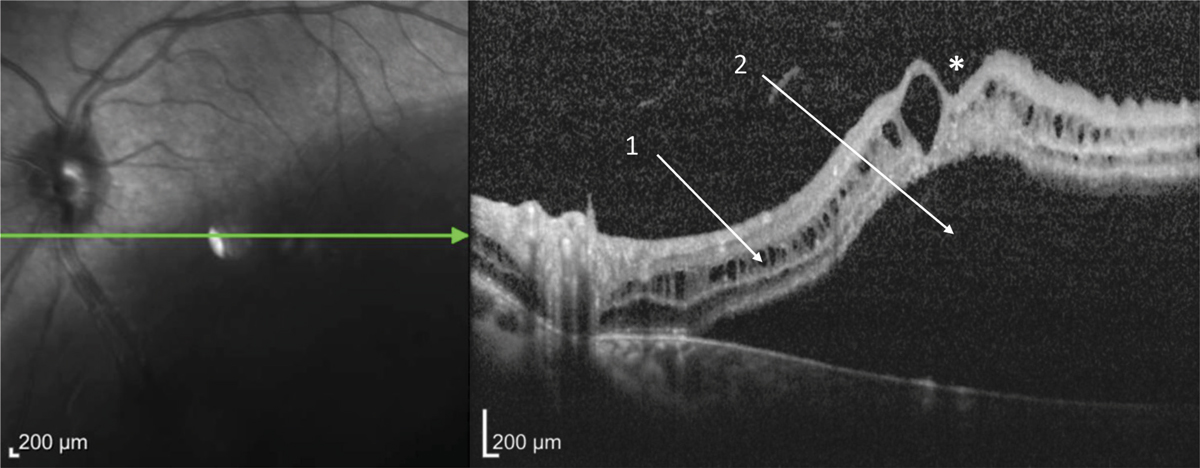
Fig. 15-22 Jeune homme de 18 ans atteint de rétinoschisis juvénile (1) compliqué de décollement de rétine (2). La macula est décollée (*).
L’angiographie à la fluorescéine peut avoir un intérêt en cas de doute avec un OMC. Aucune hyperfluorescence maculaire n’est notée en angiographie en cas de rétinoschisis, en l’absence d’atrophie maculaire associée.
L’électrorétinogramme (ERG) était un examen incontournable dans le diagnostic de la maladie avant l’avènement de l’OCT. Il garde aujourd’hui un intérêt dans les formes atypiques en particulier lorsque le profil maculaire est conservé en OCT sur l’un des deux yeux ou dans les formes évoluées compliquées d’atrophie maculaire. L’ERG montre typiquement une réduction de l’amplitude de l’onde b et une relative préservation de l’onde a avec un rapport b/a diminué. Avec l’âge et dans les formes associées à une atrophie étendue de l’épithélium pigmentaire (EP), l’onde a peut elle aussi être diminuée. Dans de rares cas, l’onde b est peu diminuée en ERG global mais elle l’est généralement en ERG multifocal.
Les modifications de l’ERG avec diminution de l’onde b ont initialement étayé l’hypothèse d’une anomalie des cellules de Müller dans le déterminisme de la maladie, d’autant plus que l’analyse histologique de la rétine de patients décédés montrait l’accumulation de matériel filamenteux apparemment issu des cellules de Müller. L’identification du gène responsable de la maladie en 1997 a permis de montrer l’implication des photorécepteurs et des cellules bipolaires et non des cellules de Müller dans le processus pathologique [9].
Le mode de transmission du rétinoschisis juvénile, mis en évidence en 1913, est récessif lié à l’X, avec une pénétrance complète et une expressivité variable. La maladie est due à des mutations du gène RS1 composé de 6 exons et localisé sur le bras court du chromosome X en position Xp22.13. Ce gène code la rétinoschisine, protéine constituée de 224 acides aminés, sécrétée par les photorécepteurs et les cellules bipolaires, qui joue un rôle crucial dans les interactions et les adhérences intercellulaires dans la rétine, en particulier l’adhérence synaptique entre les photorécepteurs et les cellules bipolaires, et possiblement un rôle dans les mouvements de fluides entre les secteurs intra- et extracellulaires [9]. En immunomarquage, la rétinoschisine est retrouvée au niveau des segments internes des photorécepteurs, des cellules bipolaires, et au niveau des plexiformes interne et externe de la rétine ; elle n’est pas retrouvée au niveau des cellules de Müller. À ce jour, plus de 200 mutations différentes du gène RS1 ont été répertoriées comme associées au rétinoschisis.
Ces mutations sont majoritairement de type faux sens et dans ce cas concernent préférentiellement les exons 4, 5 et 6 du gène qui constituent le domaine discoïdine. Les autres types de mutations intéressent toutes les régions de la protéine RS1 de façon plus homogène. Aucune corrélation significative n’a été retrouvée entre génotype et phénotype à ce jour en dehors d’une étude de Vincent et al. qui observe des anomalies moins marquées de l’ERG en cas de mutation faux sens [10].
Récemment, des essais de thérapie génique chez la souris rendue déficiente en rétinoschisine ont donné des résultats encourageants puisqu’une restauration significative de la structure et de la fonction rétinienne a pu être obtenue [9, 11].
Le diagnostic de rétinoschisis chez un jeune garçon implique un bilan ophtalmologique complet avec réfraction sous cycloplégie de façon à dépister l’hypermétropie fréquemment associée à la maladie et prescrire la correction optimale. Si une amblyopie relative existe, elle a souvent une double part, anatomique et fonctionnelle, notamment en cas de strabisme associé. Une rééducation est alors nécessaire pour agir sur la part fonctionnelle avant l’âge de 10 ans.
Des photos du fond d’œil et un examen par OCT seront systématiques pour faire un bilan de la maladie et permettre l’évaluation objective de son évolutivité au cours du temps. Un ERG est habituellement réalisé même si dans les cas typiques, il n’est pas impératif pour établir le diagnostic.
En l’absence de complications, aucun traitement n’est habituellement proposé. En effet, aucun traitement laser, ni au bord d’un rétinoschisis périphérique ni en son sein, n’empêchera la progression du rétinoschisis. Il peut éventuellement présenter un rare intérêt pour traiter une déhiscence du feuillet externe au sein d’un rétinoschisis périphérique. Plusieurs cas de DR secondaires à un traitement laser ont été décrits dans la littérature par surdosage de celui-ci entraînant l’apparition de déhiscences iatrogènes du feuillet externe [2].
La pratique de sports de combat et de sports violents (rugby, football américain, etc.) est déconseillée pour minimiser le risque d’hémorragie intravitréenne.
Le traitement préventif des complications du rétinoschisis juvénile reste aujourd’hui débattu, Yu a proposé la réalisation d’une vitrectomie systématique pour freiner l’évolution de la maculopathie et prévenir la survenue de complications dans une étude prospective non randomisée [12]. Sur une série de 17 yeux d’enfants âgés en moyenne de 12 ans, il a évalué l’efficacité de la vitrectomie (avec décollement du vitré en peropératoire, pelage de la limitante interne et tamponnement par gaz) par rapport à un groupe témoin (11 yeux) avec un recul moyen de 34,7 mois. Il a retrouvé, de façon statistiquement significative, une amélioration de l’acuité visuelle dans le groupe traité de même qu’une amélioration du profil maculaire en OCT avec disparition du schisis maculaire et un taux de complications secondaires moindre après chirurgie (12 % de DR dans le groupe traité versus 28 % dans le groupe témoin). Ces résultats restent toutefois assez isolés avec de rares publications et il n’existe actuellement aucun consensus sur la prise en charge des rétinoschisis avec maculopathie évolutive en l’absence de complications. La surveillance simple reste donc l’attitude la plus répandue.
Cette complication survient le plus souvent dans les deux premières décennies de la vie alors que le vitré n’est pas décollé. Dans la majorité des cas, la vitrectomie n’est pas nécessaire et la résorption du sang se fait sans séquelle. Une vitrectomie n’est indiquée qu’en cas d’hémorragie récidivante ou très dense sans tendance à la résorption. La vitrectomie permettra la coagulation du ou des vaisseaux responsables. Lorsque le vitré n’est pas dissécable, une ablation plus ou moins complète du feuillet interne de la rétine est parfois nécessaire pour supprimer les tractions du vitré sur la rétine [13].
Le décollement rhegmatogène est secondaire à l’existence de déhiscences présentes dans les feuillets interne et externe de la rétine au sein du schisis ou en rétine saine. Au sein du schisis périphérique, les trous au niveau du feuillet interne sont en général facilement repérables mais ne sont pas à eux seuls responsables du décollement de rétine. En revanche, la recherche des trous au niveau du feuillet externe peut s’avérer plus difficile : ils se situent le plus souvent au niveau du bord postérieur du schisis périphérique. Leur traitement est indispensable à la réapplication de la rétine. Une chirurgie ab externo peut être réalisée dans ce type de DR si la déchirure n’est pas trop postérieure ou en l’absence de prolifération vitréorétinienne. Celle-ci est une complication fréquente dans les suites avec un taux de récidive de décollement proche de 40 % [14]. Un abord endoculaire est donc souvent nécessaire.
Dans cette pathologie, les décollements de rétine tractionnels s’étendent postérieurement au rétinoschisis périphérique et sont probablement favorisés par une très grande adhérence entre le cortex vitréen et le feuillet interne du rétinoschisis périphérique. Pour ces DR, une chirurgie endoculaire est réalisée associant vitrectomie avec décollement de la hyaloïde postérieure si possible ; une rétinectomie du mur interne du schisis peut s’avérer nécessaire lorsque la hyaloïde n’est pas dissécable. Une cautérisation des vaisseaux flottants associée à un tamponnement interne par du gaz ou silicone est alors souvent nécessaire [15].
Le résultat anatomique et fonctionnel de la chirurgie du DR compliquant un rétinoschisis juvénile lié à l’X est plutôt encourageant, avec un taux de réapplication proche de 85 % [16, 17] et une amélioration de l’acuité visuelle dans 50 à 67 % des cas, mais les séries restent peu nombreuses et de faible effectif. Ces résultats doivent donc être interprétés avec réserve.
Une enquête génétique doit être proposée systématiquement à tous les sujets atteints, ainsi qu’aux possibles conducteurs et à tous les membres de la famille en général pour que le mode de transmission lié à l’X soit expliqué. En dehors d’un mariage consanguin, dans la descendance d’un homme atteint de rétinoschisis, toutes les filles seront obligatoirement conductrices et aucun des fils ne sera atteint ; dans la descendance d’une femme conductrice, le risque d’avoir un fils atteint sera de 50 % de même que celui d’avoir une fille conductrice. Du fait de la transmission avec pénétrance complète, tous les hommes porteurs de la mutation seront atteints mais de façon variée au sein d’une même famille en raison de l’expressivité variable de la mutation.
Le rétinoschisis juvénile lié à l’X peut atteindre les femmes, dans de rares cas de familles consanguines, et dans le cas encore plus rare d’association de la mutation à un syndrome de Turner (45/X). En dehors de ces circonstances exceptionnelles, les femmes conductrices sont saines et aucune anomalie ophtalmologique attribuable à la mutation n’est habituellement détectable, sur le plan fonctionnel ou anatomique. Il semble exister un consensus sur le fait que les quelques anomalies rapportées dans la littérature chez des femmes conductrices ne sont pas à rattacher à une expression même partielle de la mutation [2].
[1] Haas J. Über das Zusammenvorkommen von Veranderugen der Retina und Choroidea. Arch Augenheikd 1898 ; 37 : 343-8.
[2] Tantri A, Vrabec TR, Cu-Unjieng A, et al. X-Linked retinoschisis : a clinical and molecular genetic review. Surv Ophthalmol 2004 ; 49 : 214-30.
[3] Tanino T, Katsumi O, Hirose T. Electrophysiological similarities between two eyes with X-linked recessive retinoschisis. Doc Ophthalmo 1985 ; l60 : 149-61.
[4] George NDL, Yates JRW, Moore AT. Clinical features in affected males with X-linked retinoschisis. Arch Ophthalmol 1996 ; 114 : 274-80.
[5] Sieving PA, MacDonald IM, Chan S. X-Linked Juvenile Retinoschisis. In : Pagon RA, Adam MP, Ardinger HH, et al. Eds. GeneReviews® [Internet]. Seattle (WA) : University of Washington ; 2003 Oct 24 : 1993-2015 [updated 2014 Aug 28].
[6] Apushkin MA, Fishman GA, Rajagopalan AS. Fundus findings and longitudinal study of visual acuity loss in patients with X-linked retinoschisis. Retina 2005 ; 25 : 612-8.
[7] Yang HS, Lee JB, Yoon YH, Lee JY. Correlation between spectral-domain OCT findings and visual acuity in X-linked retinoschisis. Invest Ophthalmol Vis Sci 2014 ; 55 : 3029-36.
[8] Al-Swaina N, Nowilaty SR. Macular hole in juvenile X-linked retinoschisis. Saudi J Ophthalmol 2013 ; 27 : 283-6.
[9] Molday RS, Kellner U, Weber BH. X-linked juvenile retinoschisis : clinical diagnosis, genetic analysis, and molecular mechanisms. Prog Retin Eye Res 2012 ; 31 : 195-212.
[10] Vincent A, Robson AG, Neveu MM, et al. A phenotype–genotype correlation study of X–Linked retinoschisis. Ophthalmology 2013 ; 120 : 1454-64.
[11] Bush RA, Wei LL, Sieving PA. Convergence of Human Genetics and Animal Studies : gene therapy for X-Linked Retinoschisis. Cold Spring Harb Perspect Med 2015 ; 5 : a017368.
[12] Yu H, Li T, Luo Y, et al. Long-term outcomes of vitrectomy for progressive X-linked retinoschisis. Am J Ophthalmol 2012 ; 154 : 394-402.
[13] Ferrone PJ, Trese MT, Lewis H. Vitreoretinal surgery for complications of congenital retinoschisis. Am J Ophthalmol 1997 ; 123 : 742-7.
[14] Rosenfeld PJ, Flynn HW Jr, McDonald HR, et al. Outcomes of vitreoretinal surgery in patients with X-linked retinoschisis. Ophthalmic Surg Lasers 1998 ; 29 : 190-7.
[15] Trese MT, Ferrone PJ. The role of inner wall retinectomy in the management of juvenile retinoschisis. Graefe’s Arch Clin Exp Ophthalmol 1995 ; 233 : 706-8.
[16] George NDL, Yates JRW, Moore AT. Clinical features in affected males with X-linked retinoschisis. Arch Ophthalmol 1996 ; 114 : 274-80.
[17] Sikkink SK, Biswas S, Parry NR, et al. X-Linked retinoschisis : an update. J Med Genet 2007 ; 44 : 225-32.
I. Audo
➤ Le syndrome d’augmentation des cônes bleus se manifeste par des anomalies de la vision nocturne et par une vision plus ou moins diminuée selon la présence de kystes maculaires.
➤ Outre ces kystes, le fond d’œil montre souvent un aspect tacheté avec hypo-autofluorescence.
➤ L’ERG est l’examen clé montrant une réponse diminuée dans les conditions scotopiques, mais une réponse hyperample lors des flashes de courtes longueurs d’onde.
➤ La bestrophinopathie autosomique dominante récessive débute généralement dans l’enfance avec une acuité visuelle diminuée à l’âge adulte. Le fond d’œil montre des taches jaunâtres bien visibles en autofluorescence. L’OCT retrouve typiquement un aspect de décollement séreux rétinien. L’ERG est normal au début avant de s’altérer progressivement alors que l’électro-oculogramme (EOG) est profondément altéré.
Outre les rétinopathies pigmentaires mentionnées précédemment (voir chapitre 14.4) au cours desquelles les remaniements kystiques peuvent survenir jusqu’à près de 50 % des cas [1] ou encore le rétinoschisis lié à l’X (voir plus haut) au cours duquel les lésions maculaires sont présentes dans 100 % des cas [2], au moins deux autres hérédo-dégénérescences s’associent classiquement à la présence de kystes intrarétiniens, le syndrome d’augmentation des cônes bleus également connu sous le terme de syndrome de Goldman-Favre et la bestrophinopathie autosomique récessive. Dans ces deux pathologies, les kystes maculaires ont peu ou pas de traduction angiographique avec une meilleure documentation et un suivi des lésions par spectral-domain optical coherence tomography (SD-OCT). L’électrophysiologie, avec l’ERG global, complété par l’EOG en cas de suspicion de bestrophinopathie, est essentielle pour un diagnostic précis et montre des altérations pathognomoniques. À côté de ces formes maintenant bien caractérisées, des maculopathies kystiques récessives ou dominantes ont été décrites.
Il n’existe pas de traitement curatif pour ces pathologies même si certains rapportent l’effet des inhibiteurs de l’anhydrase carbonique qui reste à démontrer dans des études randomisées. Leur prise en charge thérapeutique inclut une correction optimale de la réfraction avec rééducation de l’amblyopie adaptée chez le jeune enfant, une protection solaire avec des verres coupant les courtes longueurs d’onde, des injections d’anti-VEGF en cas de complication néovasculaire, une prise en charge en rééducation de basse vision, un accompagnement scolaire et socioprofessionnel approprié.
Le syndrome d’augmentation des cônes bleus est aussi appelé enhanced S-cone syndrome (ESCS) ou syndrome de Goldman-Favre (mendelian inheritance in man [MIM] no 268100).
Il s’agit d’une pathologie autosomique récessive liée à des mutations sur le facteur de transcription NR2E3 [3]. La prévalence exacte de cette pathologie est inconnue mais est sûrement sous-estimée, car souvent classifiée parmi les rétinopathies pigmentaires. Elle doit cependant en être distinguée en raison de son évolutivité plus lente avec un pronostic visuel essentiellement conditionné par les modifications kystiques maculaires.
Goldmann puis Favre ont rapporté de façon indépendante les cas de frères et sœurs adolescents présentant des troubles de vision nocturne, des modifications vitréennes et un rétinoschisis central et périphérique [4, 5]. Ont suivi plusieurs autres descriptions de cas avec cécité nocturne précoce, modifications kystiques maculaires et remaniements pigmentaires [6, 7] puis la description des altérations fonctionnelles correspondant à une augmentation de la réponse des cônes sensibles aux courtes longueurs d’onde dit « cônes bleus » [8, 9].
Le syndrome d’augmentation des cônes bleus à une grande variabilité clinique [10] avec, classiquement, des troubles de la vision nocturne depuis la petite enfance, une acuité visuelle variable, conditionnée par la présence ou non de kystes maculaires, un champ visuel qui peut-être normal ou présenter des altérations concentriques corrélées aux modifications du fond d’œil. L’examen du fond d’œil est également variable d’un patient à un autre et peut être normal, surtout chez les enfants. On peut aussi trouver un aspect tacheté de l’épithélium au pôle postérieur, des modifications kystiques maculaires et des remaniements pigmentaires, plutôt chez l’adulte (distincts des ostéoblastes des rétinopathies pigmentaires, car situés au niveau de la couche de l’épithélium pigmentaire), d’aspect arrondi, nummulaire, répartis le long des arcades vasculaires. L’autofluorescence peut montrer une zone d’hypo-autofluorescence en dehors des arcades vasculaires et des modifications stellaires maculaires en présence de kystes. La prévalence de ces kystes maculaires, visibles en OCT dans la plexiforme externe ou interne, sans traduction angiographique, n’est pas connue. Un rétinoschisis périphérique peut également être présent [10]. De plus, l’aspect tacheté au fond d’œil correspond en OCT à des modifications aux dépens de la ligne ellipsoïde et d’interdigitation qui serait des rosettes formées des articles externes des photorécepteurs dysplasiques [11]. Des cas de néovaisseaux choroïdiens ainsi que de fibroses sous-rétiniennes ont été rapportés (fig. 15-23) [12, 13].
L’ERG global met en évidence des altérations fonctionnelles pathognomoniques [8, 9] : dans les conditions scotopiques, la réponse au flash de faible intensité est non discernable du bruit de fond ; la réponse à un flash standard dans les conditions scotopiques ressemble à celle au flash standard dans les conditions photopiques ; l’amplitude de la réponse au flicker 30 Hz dans les conditions photopiques, normalement comprise entre l’amplitude de l’onde a et celle de l’onde b au simple flash standard, est ici inférieure à celle de l’onde a. Enfin, la stimulation à un flash de courte longueur d’onde génère une réponse hyperample par rapport à la normale. Ces altérations témoignent de l’absence de bâtonnets fonctionnels et d’une augmentation de réponses des cônes sensibles aux courtes longueurs d’onde (fig. 15-24).
Ces altérations fonctionnelles sont en rapport avec le rôle du facteur de transcription NR2E3, impliqué dans le programme de différenciation des cônes au niveau des progéniteurs rétiniens tardifs et essentiel à la différenciation en bâtonnets [14, 15]. Ainsi, la perte de fonction de NR2E3 par des mutations entraîne une absence de différenciation des bâtonnets avec, par défaut, une différenciation des progéniteurs tardifs en cônes sensibles aux courtes longueurs d’onde (dit cônes bleus), habituellement minoritaires (8 à 10 % des cônes totaux [16]). Ceci explique les troubles de vision nocturne, l’absence de réponses fonctionnelles issues des bâtonnets et l’excès numérique et fonctionnel des cônes sensibles aux courtes longueurs d’onde. Les remaniements kystiques seraient secondaires aux altérations de l’architecture rétinienne par défaut d’adhésions intercellulaires comme dans le rétinoschisis lié à l’X. Plusieurs études suggèrent que les inhibiteurs de l’anhydrase carbonique pourraient avoir un bénéfice dans l’affaissement des kystes [17, 18] mais des études à long terme sont nécessaires pour déterminer leur effet sur l’acuité visuelle à long terme.
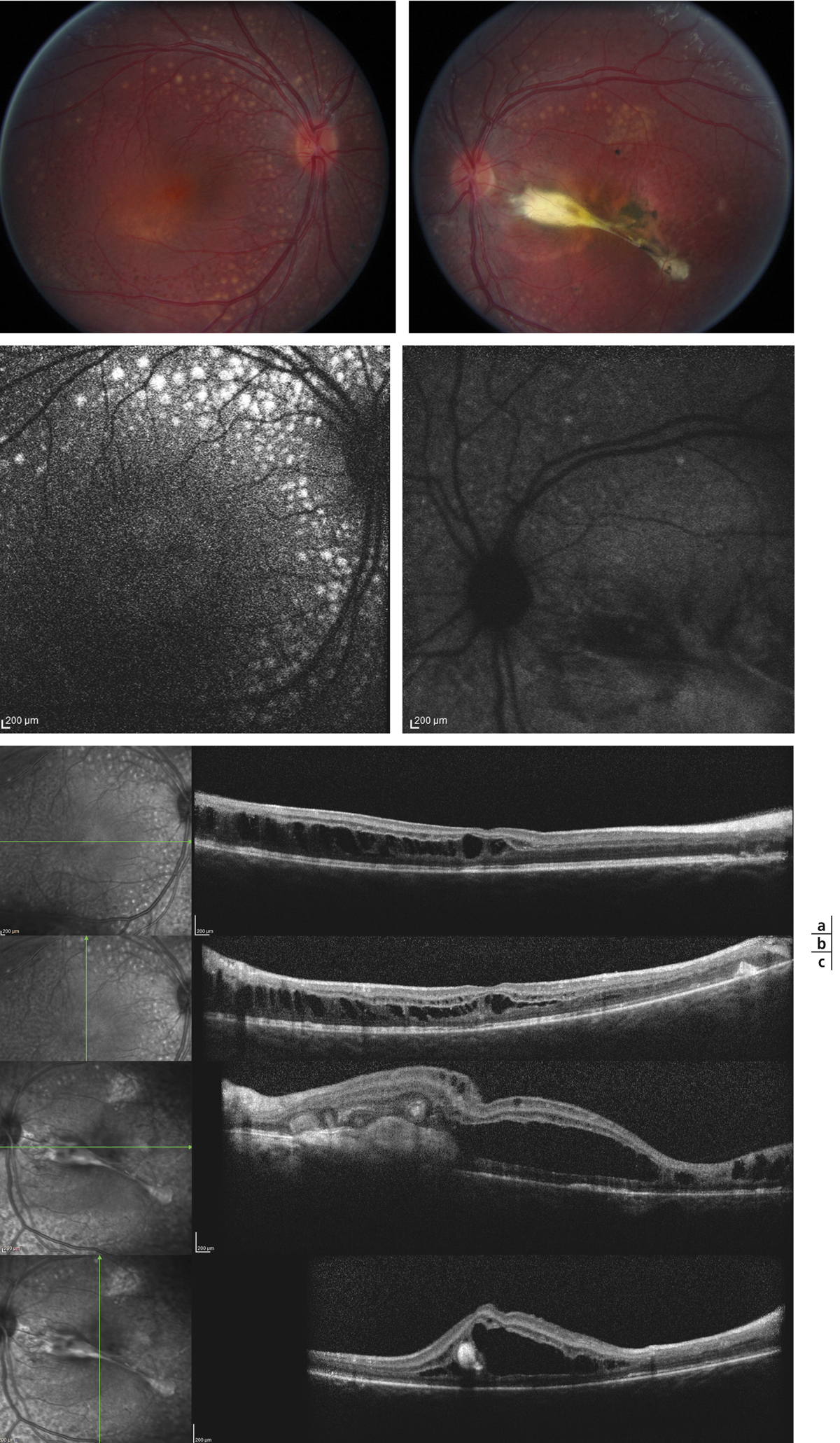
Fig. 15-23 Imagerie du fond d’œil d’un jeune homme de 12 ans atteint de syndrome d’augmentation des cônes bleus associé à une mutation homozygote (c.932G>A ; p. Arg311Gln) sur le gène NR2E3.
a. Rétinophotographies révélant de multiples taches jaunâtres en moyenne périphérie avec des remaniements kystiques maculaires ; à gauche on note une fibrose sous-rétinienne maculaire. b. Autofluorescence du fond de l’œil qui révèle de multiples taches hyper-autofluorescentes correspondant à une lésion jaunâtre du fond de l’œil ; on note également des modifications de l’autofluorescence fovéolaire en rapport avec des modifications kystiques maculaires et la fibrose sous-rétinienne à l’œil gauche. c. Le SD-OCT révèle des modifications kystiques maculaires au sein des couches nucléaires externe et interne non centrées sur la fovéa.
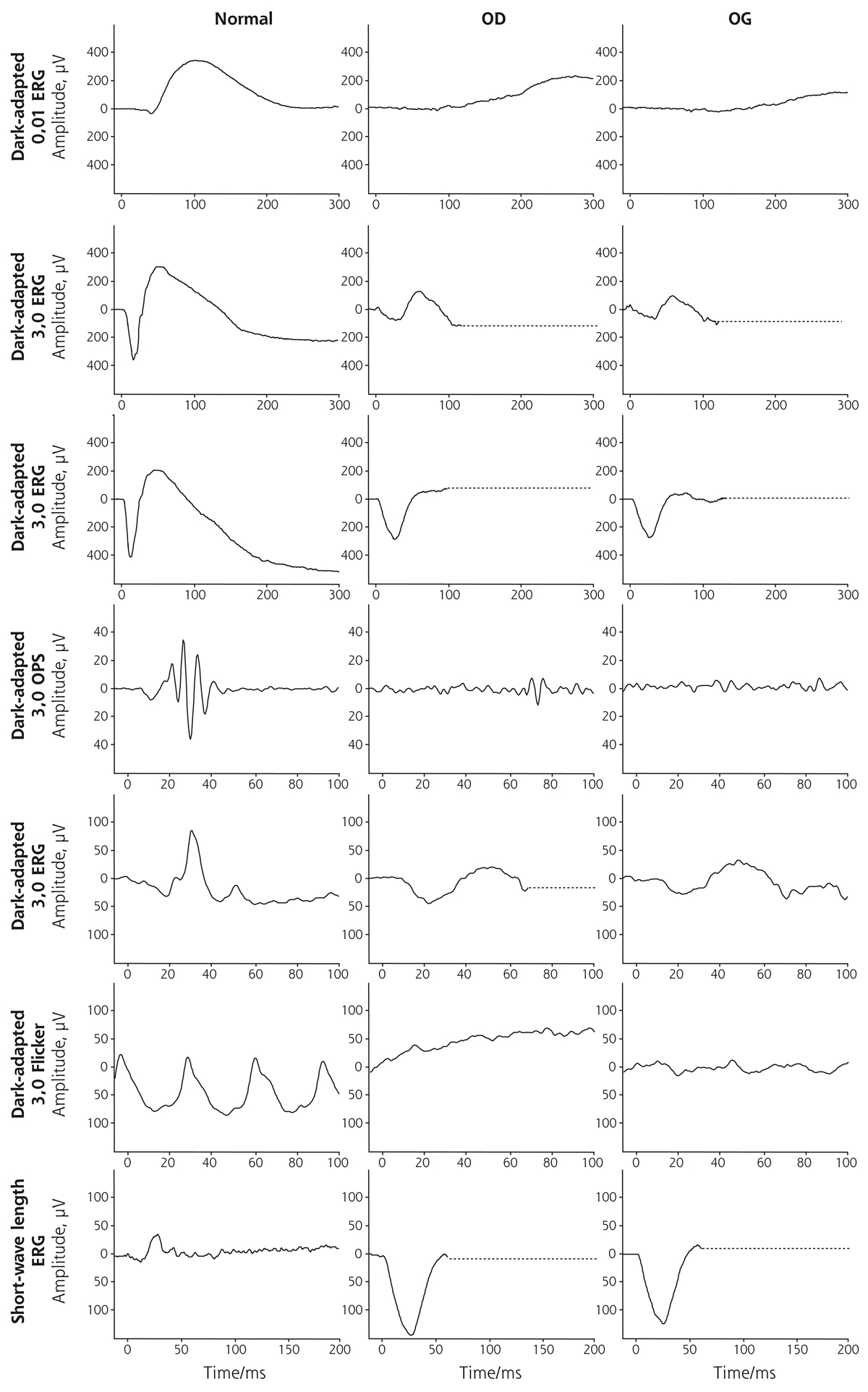
Fig. 15-24 Altérations fonctionnelles du syndrome d’augmentation des cônes bleus à l’électrorétinogramme champ total.
Dans les conditions scotopiques, la réponse au flash de faible intensité (dark adapted 0.01 ERG) est non discernable du bruit de fond ; la réponse à un flash standard dans les conditions scotopiques (dark adapted 3.0 ERG) ressemble à celle au flash standard dans les conditions photopiques (light adapted 3.0 ERG) ; l’amplitude de la réponse au flicker 30 Hz (light adapted 3.0 Flicker) dans les conditions photopiques, normalement comprise entre l’amplitude de l’onde a et celle de l’onde b au simple flash standard, est ici inférieure à celle de l’onde a. Enfin, la stimulation à un flash de courte longueur d’onde (short wave length) génère une réponse hyperample par rapport à la normale. Ces altérations témoignent de l’absence de bâtonnets fonctionnels et d’une augmentation de réponses des cônes sensibles aux courtes longueurs d’onde.
Récemment, une nouvelle entité associée à des mutations autosomiques récessives sur le gène BEST1 a été décrite sous le nom de bestrophinopathie autosomique récessive [19], des mutations sur le même gène ayant été impliquées antérieurement dans au moins trois autres pathologies autosomiques dominantes : maculopathie vitelliforme de Best (MIM no 608161) ; maculopathie pseudovitelliforme de l’adulte ; vitréochoroïdopathie autosomique dominante (MIM no 193220). La bestrophinopathie autosomique récessive est associée à des mutations entraînant une perte de fonction de BEST1 contrairement à la dystrophie vitelliforme de Best associée à des mutations à effet dominant négatif.
L’âge de début est variable mais typiquement dans l’enfance. Le tableau clinique inclut une hypermétropie avec courte longueur axiale et risques de glaucome par fermeture de l’angle nécessitant un traitement préventif spécifique [19, 20]. L’acuité visuelle est variable : relativement conservée chez les enfants, elle diminue à l’âge adulte jusqu’à 1/10 à « compte les doigts ». Le champ visuel retrouve un scotome central avec préservation des isoptères périphériques et la vision des couleurs montre de façon non spécifique des degrés variables de dyschromatopsie d’axe bleu-jaune. L’électrorétinogramme global est normal chez les enfants et s’altère progressivement sur les réponses scotopiques et photopiques contrairement à la maculopathie vitelliforme de Best [21]. Par contre, comme dans toutes les pathologies associées à des mutations sur BEST1, l’électro-oculogramme est profondément altéré, au-delà des conséquences de l’altération de la fonction des photorécepteurs sur cet examen avec des rapports d’Arden proches de 100 %.
L’examen du fond d’œil met en évidence de grossières taches jaunâtres au pôle postérieur avec des remaniements maculaires kystiques. Les dépôts jaunâtres sont mieux visibles par imagerie en autofluorescence laquelle souligne aussi l’aspect stellaire des remaniements maculaires kystiques. L’image typique en OCT révèle un décollement séreux rétinien qui peut concerner l’ensemble du pôle postérieur. À celui-ci, dans les formes débutantes, s’associe un aspect d’allongement de la ligne ellipsoïde et d’interdigitation qui représenterait un allongement des articles externes des photorécepteurs, et la présence de kystes intrarétiniens dans les couches nucléaires externe et interne. Les lésions jaunâtres correspondent à des dépôts hyper-réflectifs entre l’épithélium et la neurorétine et semblent similaires aux dépôts vitellins de Best dominant (fig. 15-25). Ces kystes n’ont pas de traduction angiographique. Dans les formes évoluées, le décollement séreux persiste mais les structures issues des articles externes des photorécepteurs disparaissent de même que les kystes intrarétiniens (fig. 15-26) [22]. Des cas de complications néovasculaires ont été rapportés avec une bonne réponse aux injections intravitréennes d’anti-VEGF [23]. Les diagnostics différentiels de cette pathologie incluent : la choriorétinite séreuse centrale, une choroïdite multifocale, une poussée de maladie de Vogt-Koyanagi-Harada, une maculopathie vitelliforme exsudative polymorphe, une maladie de Best multifocale, une maculopathie de Stargardt ou une dystrophie réticulée [24]. Des tests électrophysiologiques ainsi que la génétique moléculaire sont essentiels pour un diagnostic précis.
L’effet des inhibiteurs de l’anhydrase carboniques semble variable avec quelques rapports d’efficacité per os ou en topique [24] mais aucune étude standardisée n’a pas encore été menée [22].
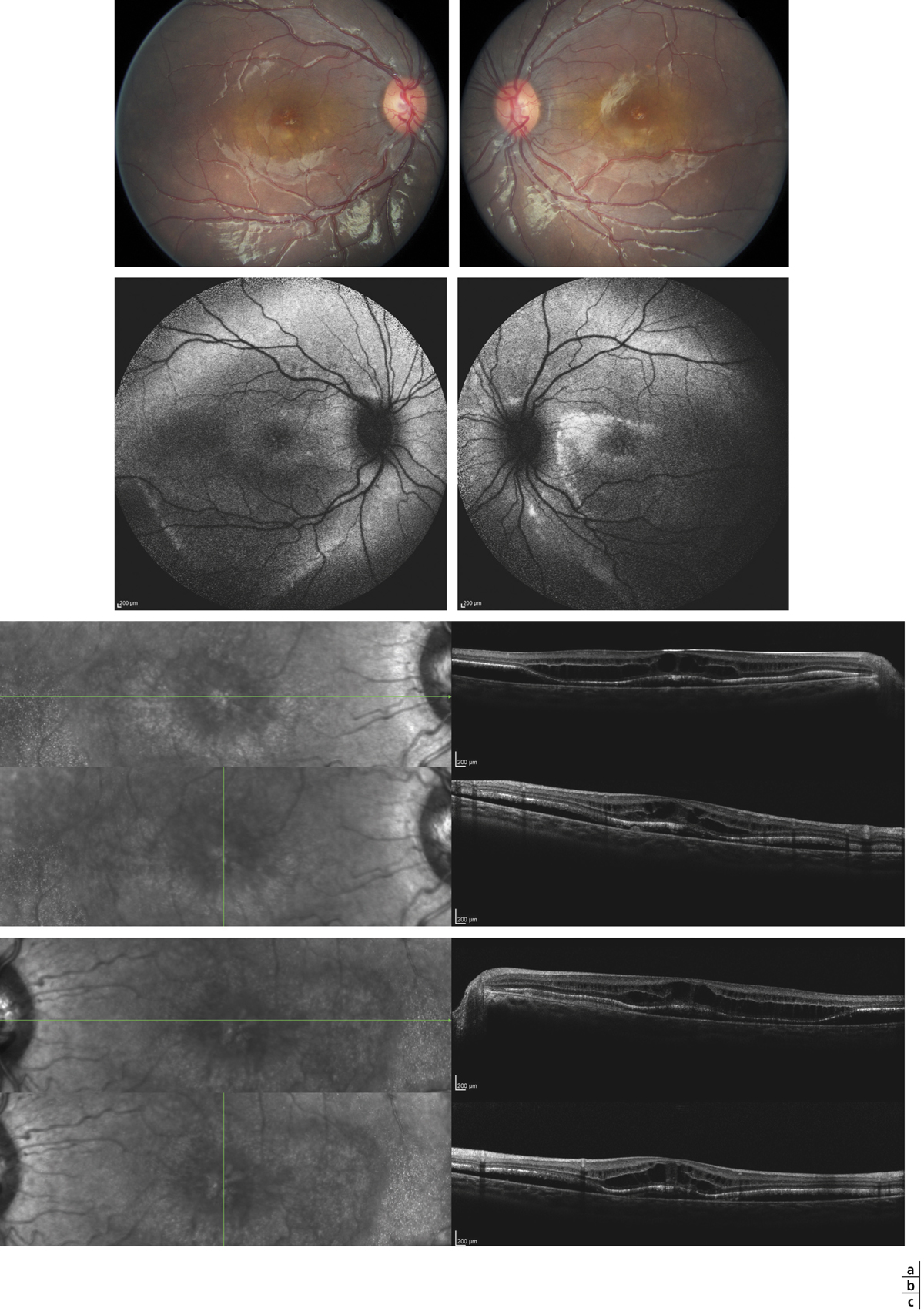
Fig. 15-25 Imagerie rétinienne d’un jeune garçon de 10 ans atteint de bestrophinopathie récessive avec confirmation moléculaire.
a, b. Les rétinophotographies (a) révèlent des taches jaunâtres grossières réparties sur le pôle postérieur qui apparaissent hyper-autofluorescentes (b). c. Le SD-OCT retrouve de nombreux kystes intrarétiniens principalement dans les couches nucléaires externe et interne, relativement centrés par rapport à la fovéa ; on note par ailleurs un décollement séreux rétinien avec un épaississement des structures des articles externes des photorécepteurs.
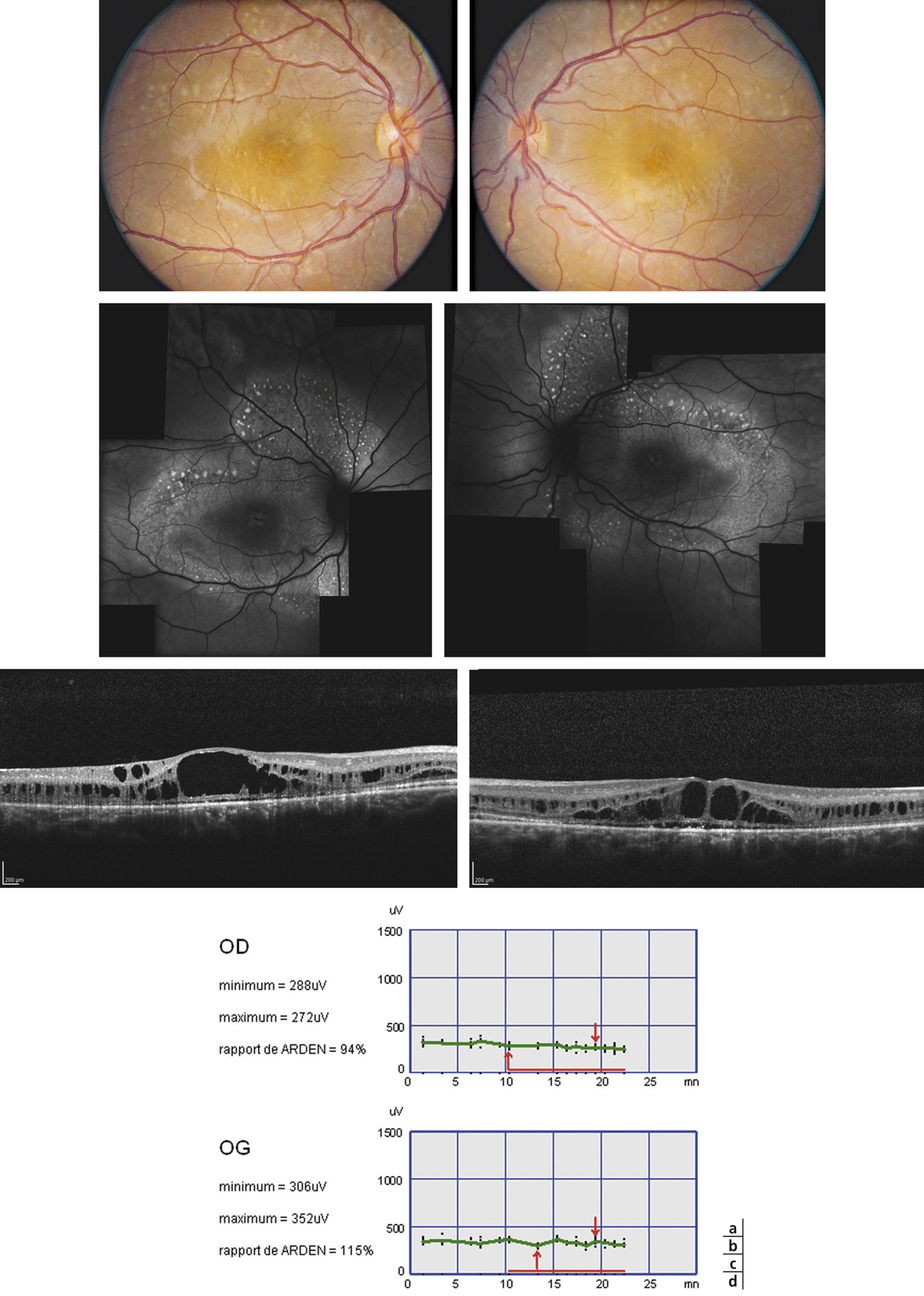
Fig. 15-26 Imagerie rétinienne d’une femme de 35 ans atteinte de bestrophinopathie récessive avec confirmation moléculaire.
Comme le cas présenté à la figure 15-25, les rétinophotographies (a) révèlent des taches jaunâtres grossières réparties sur le pôle postérieur qui apparaissent hyper-autofluorescentes (b), ainsi que des modifications kystiques maculaires (c). Cependant, chez cette patiente, le décollement séreux persiste mais les structures correspondant aux articles externes des photorécepteurs ont disparu. Aspect de l’EOG correspondant (d).
Quelques rares cas d’œdème maculaire cystoïde autosomique dominant ont été rapportés tel l’exemple d’une grande famille hollandaise récemment revisitée [25] avec comme signes distinctifs une hypermétropie avec quelques cas de glaucome par fermeture de l’angle, un œdème maculaire avec hyperfluorescence en angiographie et une fonction rétinienne (ERG et EOG) conservée dans les stades précoces, une dégénérescence rétinienne progressive associée à une dysfonction de l’épithélium pigmentaire puis des photorécepteurs lors de l’évolution. La cause génétique reste inconnue.
[1] Adackapara CA, Sunness JS, Dibernardo CW, et al. Prevalence of cystoid macular edema and stability in oct retinal thickness in eyes with retinitis pigmentosa during a 48-week lutein trial. Retina 2008 ; 28 : 103-10.
[2] Yu J, Ni Y, Keane PA, et al. Foveomacular schisis in juvenile X-linked retinoschisis : an optical coherence tomography study. Am J Ophthalmol 2010 ; 149 : 973-8 e2.
[3] Haider NB, Jacobson SG, Cideciyan AV, et al. Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nature genetics 2000 ; 24 : 127-31.
[4] Goldmann H. Biomicroscopie du corps vitré et du fond de l’œil. Bull Mem Soc Fr Ophthalmol 1957 ; 70 : 265-72.
[5] Favre M. Two cases of hyaloid-retinal degeneration. Ophthalmologica 1958 ; 135 : 604-9.
[6] Ricci A. Clinique et transmission génétique des différentes formes de dégénérescences vitréo-rétiniennes. Ophthalmologica 1960 ; 139 : 338-42.
[7] MacVicar JE, Wilbrandt HR. Hereditary retinoschisis and early hemeralopia. A report of two cases. Archives of Ophthalmology 1970 ; 83 : 629-36.
[8] Jacobson SG, Marmor MF, Kemp CM, Knighton RW. SWS (blue) cone hypersensitivity in a newly identified retinal degeneration. Investigative Ophthalmology & Visual Science 1990 ; 31 : 827-38.
[9] Marmor MF, Jacobson SG, Foerster MH, et al. Diagnostic clinical findings of a new syndrome with night blindness, maculopathy, and enhanced S cone sensitivity. Am J Ophthalmol 1990 ; 110 : 124-34.
[10] Audo I, Michaelides M, Robson AG, et al. Phenotypic variation in enhanced S-cone syndrome. Investigative Ophthalmology & Visual Science 2008 ; 49 : 2082-93.
[11] Wang NK, Fine HF, Chang S, et al. Cellular origin of fundus autofluorescence in patients and mice with a defective NR2E3 gene. Br J Ophthalmol 2009 ; 93 : 1234-40.
[12] Khan AO, Aldahmesh MA, Al-Harthi E, Alkuraya FS. Helicoid subretinal fibrosis associated with a novel recessive NR2E3 mutation p.S44X. Archives of Ophthalmology 2010 ; 128 : 344-8.
[13] Yzer S, Barbazetto I, Allikmets R, et al. Expanded clinical spectrum of enhanced S-cone syndrome. JAMA Ophthalmology 2013 ; 131 : 1324-30.
[14] Peng GH, Ahmad O, Ahmad F, et al. The photoreceptor-specific nuclear receptor Nr2e3 interacts with Crx and exerts opposing effects on the transcription of rod versus cone genes. Human Molecular Genetics 2005 ; 14 : 747-64.
[15] Chen J, Rattner A, Nathans J. The rod photoreceptor-specific nuclear receptor Nr2e3 represses transcription of multiple cone-specific genes. J Neurosci 2005 ; 25 : 118-29.
[16] Ahnelt PK. The photoreceptor mosaic. Eye 1998 ; 12 : 531-40.
[17] Thompson DA, Ali RR, Banin E, et al. Advancing therapeutic strategies for inherited retinal degeneration : recommendations from the Monaciano Symposium. Investigative Ophthalmology & Visual Science 2015 ; 56 : 918-31.
[18] Genead MA, Fishman GA, McAnany JJ. Efficacy of topical dorzolamide for treatment of cystic macular lesions in a patient with enhanced S-cone syndrome. Documenta Ophthalmologica Advances in Ophthalmology 2010 ; 121 : 231-40.
[19] Burgess R, Millar ID, Leroy BP, et al. Biallelic mutation of BEST1 causes a distinct retinopathy in humans. American Journal of Human Genetics 2008 ; 82 : 19-31.
[20] Crowley C, Paterson R, Lamey T, et al. Autosomal recessive bestrophinopathy associated with angle-closure glaucoma. Documenta Ophthalmologica Advances in Ophthalmology 2014 ; 129 : 57-63.
[21] Borman AD, Davidson AE, O’Sullivan J, et al. Childhood-onset autosomal recessive bestrophinopathy. Archives of Ophthalmology 2011 ; 129 : 1088-93.
[22] Fung AT, Yzer S, Goldberg N, et al. New best1 mutations in autosomal recessive bestrophinopathy. Retina 2015 ; 35 : 773-82.
[23] Iannaccone A, Kerr NC, Kinnick TR, et al. Autosomal recessive best vitelliform macular dystrophy : report of a family and management of early-onset neovascular complications. Archives of Ophthalmology 2011 ; 129 : 211-7.
[24] Boon CJ, van den Born LI, Visser L, et al. Autosomal recessive bestrophinopathy : differential diagnosis and treatment options. Ophthalmology 2013 ; 120 : 809-20.
[25] Saksens NT, van Huet RA, van Lith-Verhoeven JJ, et al. Dominant cystoid macular dystrophy. Ophthalmology 2015 ; 122 : 180-91.
J.-B. Conart
➤ La fossette colobomateuse (FC) est une affection congénitale de la papille qui se complique d’un décollement séreux maculaire dans 25 à 75 % des cas.
➤ Si une résorption spontanée est possible, le traitement s’impose en cas de baisse de vision prolongée en raison du risque de survenue d’une atrophie de l’épithélium pigmentaire.
➤ Son but est de créer une cicatrice choriorétinienne étanche empêchant la diffusion de liquide entre la fossette et l’espace sous-rétinien et de relâcher les tractions du vitré sur la rétine juxtapapillaire.
➤ Malgré l’absence de consensus, la vitrectomie associée à une photocoagulation laser et à un tamponnement par gaz semble offrir les meilleurs résultats anatomiques et fonctionnels.
La fossette colobomateuse (FC) de la papille est une affection congénitale rare qui atteint une personne pour onze mille dans la population générale [1]. Elle a été décrite pour la première fois par Wiethe en 1882 [2] et serait liée à un défaut de fermeture de la fente embryonnaire [1, 3]. Elle survient le plus souvent de façon sporadique et touche également les deux sexes [1]. Unilatérale dans 85 à 90 % des cas, elle se situe le plus souvent dans la partie inférotemporale de la papille [1, 3, 4]. Cette affection, habituellement asymptomatique en l’absence de complication, est généralement découverte au décours d’un examen systématique du fond d’œil. Elle peut se compliquer dans 25 à 75 % des cas d’un décollement séreux rétinien maculaire responsable d’une baisse de vision, survenant le plus souvent entre l’âge de 30 et 40 ans [1].
Plusieurs barrières anatomiques bloquent normalement la libre circulation du liquide cérébrospinal au niveau de la papille :la barrière hémato-encéphalique, la lame criblée du nerf optique, le tissu intermédiaire de Kuhnt et les jonctions serrées de l’épithélium pigmentaire (EP) de la rétine [5]. Aucune barrière ne s’oppose en revanche à la libre circulation du liquide d’origine vitréenne (fig. 15-27a).
L’origine du liquide contenu dans le décollement séreux rétinien reste controversée avec trois théories :
l’origine vitréenne du liquide a été évoquée par des études histopathologiques qui ont montré la présence de mucopolysaccharides vitréens au sein de FC [6]. Le passage de gaz ou d’huile de silicone intra-oculaire sous la rétine à travers la fossette ou la possibilité de drainer le liquide sous-rétinien par aspiration devant la fossette étayent cette hypothèse [3, 7] ;
l’origine cérébrospinale a été proposée par Gass qui a constaté une faiblesse des barrières naturelles au niveau de la fossette [8]. Sur le plan histologique, la FC est en effet une hernie de rétine dysplasique qui s’étend en arrière dans l’espace sous-arachnoïdien à travers une déhiscence de la lame criblée [6] ;
une origine suprachoroïdienne du liquide avec diffusion à partir des vaisseaux choroïdiens semble moins probable.
En 1988, Lincoff émet l’hypothèse, sur la base de constatations biomicroscopiques, que le liquide issu de la fossette commence par infiltrer la couche des fibres optiques puis crée une séparation pseudo-schisique des couches internes de la rétine [9]. Le décollement séreux rétinien ne serait donc qu’une conséquence de ce schisis intrarétinien, le liquide accédant secondairement à l’espace sous-rétinien à partir d’une déhiscence située dans les couches externes [1, 9]. Le vitré semble également jouer un rôle important dans la pathogénie du décollement maculaire avec régression spontanée du décollement séreux rétinien et des phénomènes angiographiques chez des patients ayant réalisé leur décollement postérieur du vitré [7]. Plusieurs auteurs ont par ailleurs rapporté la présence d’une déhiscence dans le tissu fibroglial recouvrant le toit de la fossette, autorisant le passage de vitré liquéfié dans l’espace sous-rétinien [1]. Cette théorie a été étayée par l’efficacité des traitements visant à diminuer ou supprimer les tractions du vitré sur la rétine juxtapapillaire et/ou maculaire : injection intravitréenne de gaz, vitrectomie voire indentation maculaire [3].
Finalement, le décollement séreux rétinien maculaire serait lié à un mécanisme mixte, tractionnel et rhegmatogène [5] : la maladie débuterait par une exagération du passage transpapillaire de divers fluides (cérébrospinal, issu de la choriocapillaire) en raison de l’anomalie des barrières (fig. 15-27b). Initialement pompé par les cellules de l’épithélium pigmentaire, le liquide sous-rétinien finirait par s’accumuler sous la rétine lorsque la capacité d’absorption des cellules pigmentaires est dépassée. L’anomalie des barrières entraîne également un passage de liquide dans la cavité vitréenne entraînant une liquéfaction précoce du vitré puis des tractions dans l’aire papillaire et enfin la formation d’une déhiscence sur le toit ou le bord de la fossette.
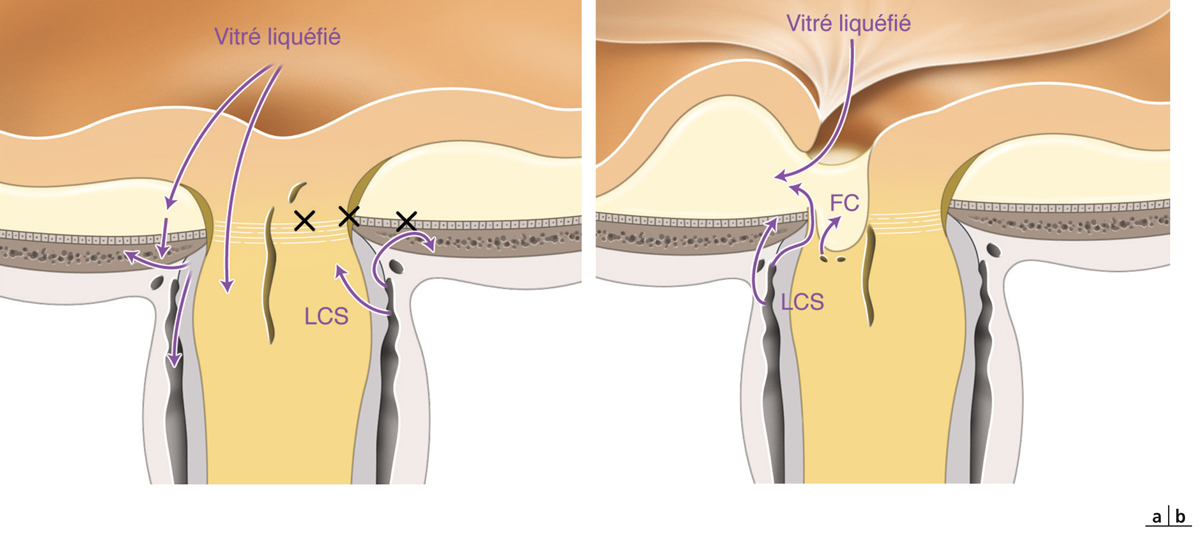
Fig. 15-27 Rappel anatomique.
a. Barrières anatomiques au niveau de la tête du nerf optique. b. Disparition des barrières anatomiques en cas de fossette colobomateuse.
FC : fossette colobomateuse ; LCS : liquide cérébrospinal.
La FC apparaît sous la forme d’une dépression focale grise, blanche ou jaune le plus souvent localisée dans la partie inférotemporale de la papille (fig. 15-28a) [1, 4]. Cette variation de couleur serait liée à la présence d’un tissu fibroglial recouvrant le toit de la fossette. La FC peut être isolée ou associée à un colobome irien ou choriorétinien (fig. 15-28b).
Habituellement asymptomatique en l’absence de complications, la FC peut parfois être responsable d’altérations du champ visuel par atteinte des fibres nerveuses [4]. La principale cause de baisse de vision est liée à la survenue dans 25 à 75 % des cas d’une infiltration rétinienne visible sous la forme d’un décollement séreux rétinien maculaire parfois associé à une dégénérescence kystique de la rétine ou à une déhiscence localisée dans les couches externes (fig. 15-29) [1, 3]. Des remaniements de l’EP consécutifs à une absorption accrue de liquide sous-rétinien peuvent apparaître secondairement et entraîner une baisse de vision persistante même si une résorption spontanée peut survenir dans environ 25 % des cas [1].
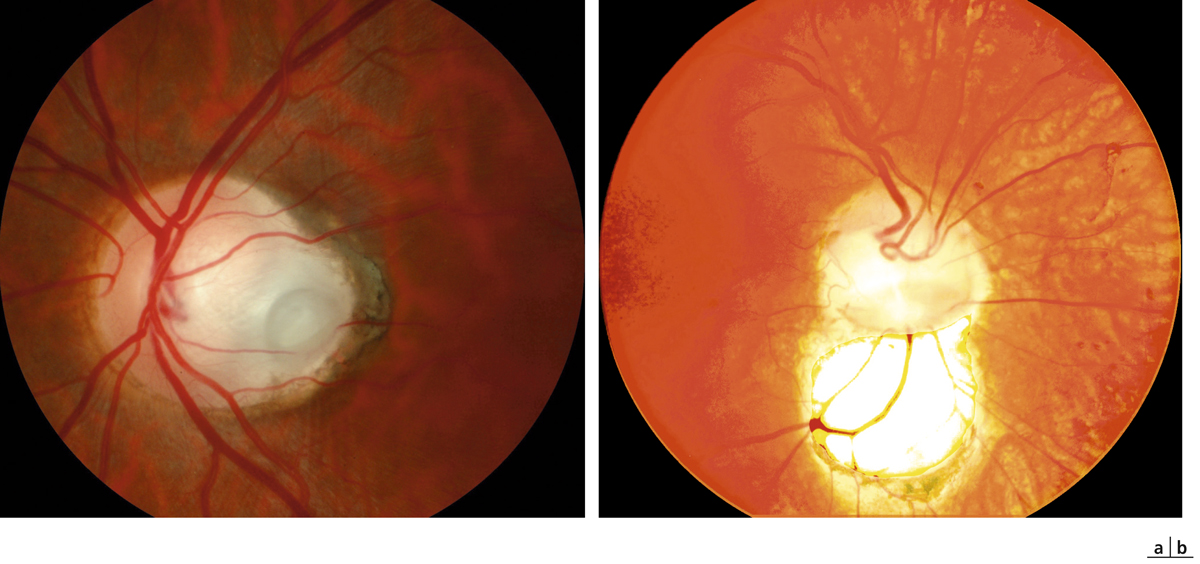
Fig. 15-28 Fossette colobomateuse de la papille.
a. Isolée. b. Associée à un colobome choriorétinien.
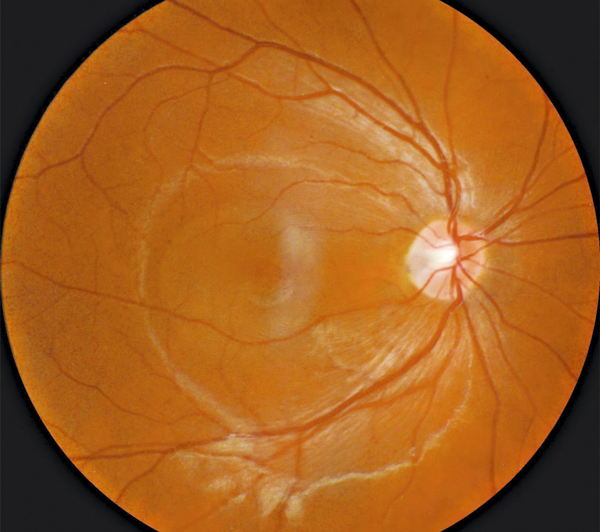
Fig. 15-29 Décollement maculaire compliquant une fossette colobomateuse.
L’OCT constitue le principal examen dans le diagnostic et le suivi des FC compliquées de décollement séreux rétinien. Le concept de structure bilaminaire proposé par Lincoff (fig. 15-30) [9] a été confirmé par plusieurs études montrant l’existence d’un espace schisique dans la rétine interne reliant directement la fossette au soulèvement maculaire (fig. 15-31 et 15-32a) [1, 10] : le fluide intrarétinien accéderait secondairement à l’espace sous-rétinien à travers une déhiscence située dans les couches externes, parfois visible à l’OCT [3, 11]. Des études plus récentes utilisant le SD-OCT ont montré que le liquide issu de la fossette pouvait pénétrer et s’accumuler dans différentes couches de la rétine [11, 12]. Imamura et al. ont ainsi observé, sur les 16 patients de leur série, que le fluide s’accumulait principalement dans la couche nucléaire externe (94 %), puis dans la couche nucléaire interne (81 %), la couche des cellules ganglionnaires (44 %) et sous la membrane limitante interne (13 %) [11]. Un décollement séreux rétinien était retrouvé chez 69 % des patients et associé à une déhiscence dans les couches externes dans 27 % des cas. Pour Roy et al., les couches externes de la rétine sont systématiquement atteintes et représentent le point de départ du fluide intrarétinien vers les autres couches [12].
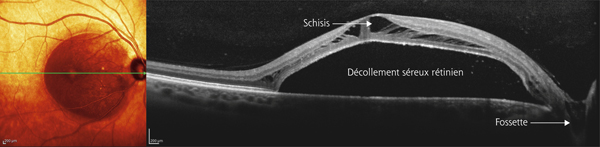
Fig. 15-30 Soulèvement bilaminaire (SD-OCT) : schisis des couches internes ; soulèvement séreux rétinien.
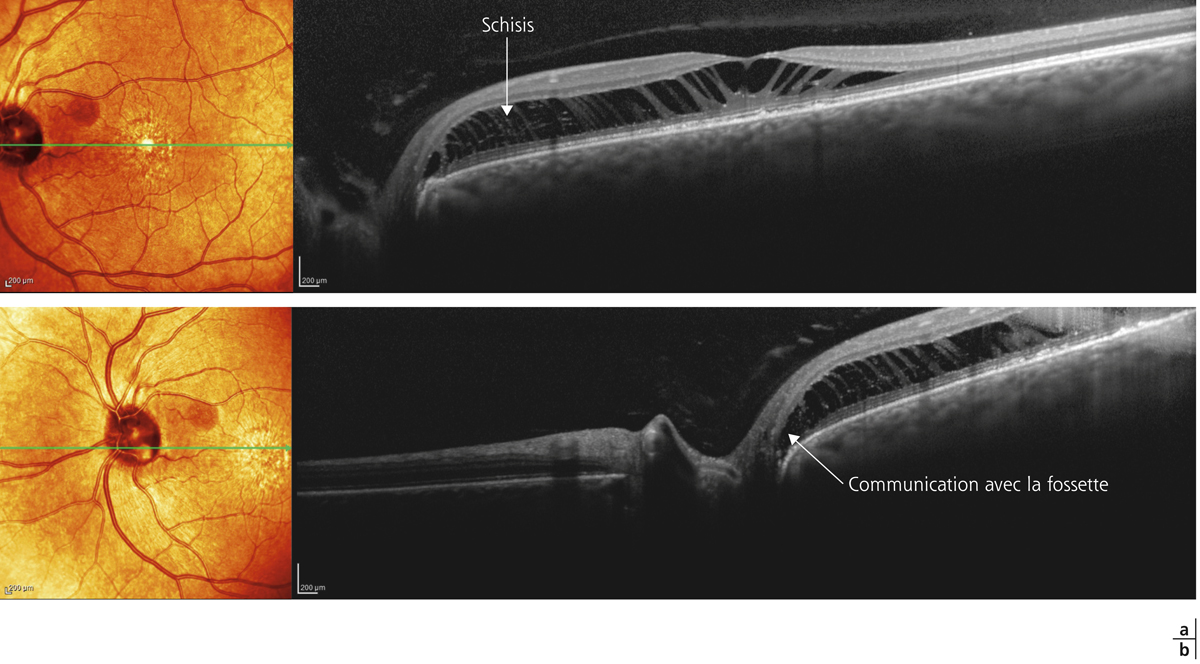
Fig. 15-31 Séparation schisique intrarétinienne (SD-OCT).
a. On ne note pas de décollement séreux rétinien associé. b. Fin espace schisique reliant le liquide intrarétinien à la fossette colobomateuse
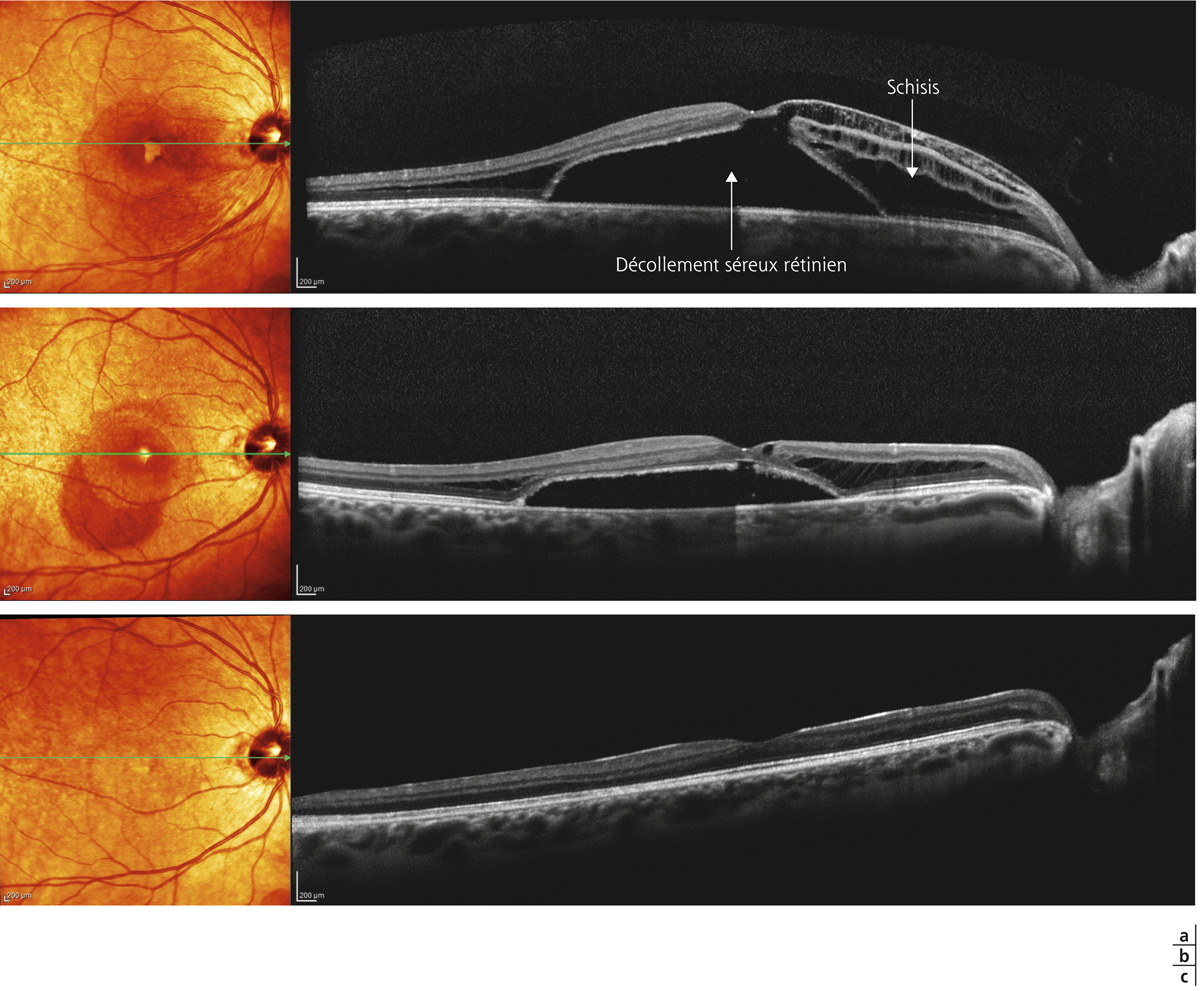
Fig. 15-32 Évolution postopératoire d’un patient traité par vitrectomie–gaz (SD-OCT).
a. Avant chirurgie, soulèvement bilaminaire. b. À 3 mois postopératoires, régression du décollement séreux rétinien et du schisis. c. À 6 mois postopératoires, résorption complète du liquide intra- et sous-rétinien.
La prise en charge des FC compliquées de décollement séreux rétinien ne fait pas l’objet d’un consensus. De nombreuses thérapeutiques ont été proposées comprenant : une photocoagulation laser seule ou associée à une injection intravitréenne de gaz, une vitrectomie ou la mise en place d’une indentation maculaire [1, 3, 5]. Si une résorption spontanée du liquide sous-rétinien est observée dans 25 % des cas, le traitement s’impose en cas de baisse de vision prolongée en raison du risque de survenue à long terme d’une dégénérescence kystique de la rétine ou d’une atrophie de l’EP limitant la récupération fonctionnelle [1]. Le but est de créer une barrière empêchant tout passage de liquide entre la fossette et l’espace sous-rétinien et de diminuer les tractions du vitré sur la rétine juxtapapillaire. Les principaux traitements et leurs résultats sont rapportés dans le tableau 15.1.
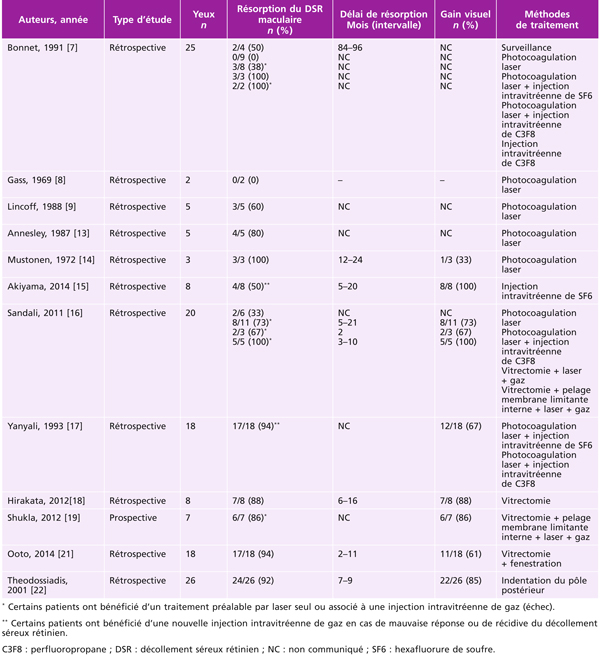
Tableau 15-1 Décollement séreux rétinien compliquant une fossette colobomateuse de la papille : principaux traitements utilisés avec leurs résultats anatomiques.
En 1969, Gass propose l’utilisation d’une photocoagulation laser du bord temporal de la papille pour créer une cicatrice choriorétinienne étanche isolant la fossette de l’espace sous-rétinien [8]. Les premiers résultats sont toutefois décevants puisqu’il n’obtient aucune réapplication chez les deux patients. Plusieurs séries sont par la suite publiées avec une efficacité variant de 0 à 100 % [7, 9, 13, 14] ! Par ailleurs le succès anatomique ne s’accompagne pas toujours d’une amélioration fonctionnelle également liée à l’état rétinien sous-jacent [14]. La résorption du liquide sous-rétinien est souvent lente et peut durer plusieurs mois. Le laser ne s’accompagne pas d’altération du champ visuel car les fibres nerveuses sont préservées si la mise au point se fait sur les plans profonds. La technique consiste à appliquer plusieurs rangées d’impacts confluents en temporal de la papille, dans la zone de soulèvement, avec une puissance suffisante pour obtenir des impacts blanc-jaune [5].
En 1998, Lincoff décrit le déplacement du liquide sous-rétinien en dehors de la région maculaire, après injection d’une bulle de gaz dans la cavité vitréenne, la rétine se réappliquant progressivement par glissement du liquide vers le bas [10]. L’efficacité est toutefois temporaire probablement du fait de la persistance d’une séparation schisique des couches internes de la rétine permettant le passage secondaire de fluide dans l’espace sous-rétinien. Akiyama et al. ont constaté un succès sans récidive dans quatre cas sur huit après injection intravitréenne de 0,3 mL d’hexafluorure de soufre (SF6) pur suivi d’un positionnement pendant 5 jours [15].
Plusieurs auteurs ont associé les trois traitements ci-dessus avec photocoagulation laser, tamponnement par gaz pour créer une barrière étanche, déplacer le liquide sous la rétine en dehors de la macula et agir sur la part rhegmatogène du décollement. En 1991, Bonnet constate une régression complète du décollement séreux rétinien dans trois cas sur huit après injection d’hexafluorure de soufre (SF6) et dans trois cas sur trois après injection de perfluoropropane (C3F8), suggérant une légère supériorité des gaz à durée de vie plus longue [7]. Cette technique permet d’obtenir un succès anatomique dans plus de trois quarts des cas [16, 17]. Le laser est réalisé avant ou après l’injection d’une bulle de gaz dans la cavité vitréenne, suivie d’un positionnement face vers le sol de durée variable selon le type de gaz utilisé.
L’efficacité relative des techniques précédemment décrites et l’implication présumée des tractions du vitré dans la survenue et les récidives du décollement séreux rétinien ont conduit à proposer la vitrectomie en première intention pour le traitement des FC compliquées. La vitrectomie est complète, associée ou non à un pelage de la membrane limitante interne (MLI) maculaire [1, 3, 18, 19]. Les taux de réapplication sont bons même si le pelage de la MLI reste controversé en raison du risque de survenue d’un trou maculaire en postopératoire [19].
D’autres auteurs préconisent d’associer la vitrectomie à une photocoagulation laser et à un tamponnement par gaz avec des résultats très satisfaisants (80 à 100 % de succès) (voir fig. 15-32 et eCas clinique 15.1 ) [3, 16, 19]. Le laser faciliterait en effet la résorption du liquide sous-rétinien en stimulant les cellules de l’épithélium pigmentaire et diminuerait les risques de récidive après vitrectomie [5]. La bulle de gaz, quant à elle, permet de déplacer le liquide en dehors de la région maculaire et agit sur la composante rhegmatogène du décollement, favorisant la formation d’une barrière étanche. Spaide et al. ont également suggéré de réaliser une fenestration dans les couches internes de la rétine juxtapapillaire afin de favoriser la migration du fluide intrarétinien dans la cavité vitréenne même si son effet reste débattu [20, 21].
) [3, 16, 19]. Le laser faciliterait en effet la résorption du liquide sous-rétinien en stimulant les cellules de l’épithélium pigmentaire et diminuerait les risques de récidive après vitrectomie [5]. La bulle de gaz, quant à elle, permet de déplacer le liquide en dehors de la région maculaire et agit sur la composante rhegmatogène du décollement, favorisant la formation d’une barrière étanche. Spaide et al. ont également suggéré de réaliser une fenestration dans les couches internes de la rétine juxtapapillaire afin de favoriser la migration du fluide intrarétinien dans la cavité vitréenne même si son effet reste débattu [20, 21].
Il faut souligner que, dans tous les cas, la résorption du liquide intra- et sous-rétinien est longue et peut prendre plusieurs mois. La nécessité du laser et du tamponnement par gaz est discutée ; Hirakata et al. ont publié une série de patients opérés par vitrectomie sans laser, ni tamponnement par gaz, avec un taux de succès égal à 90 %, et une réapplication rétinienne complète après un délai moyen de 12 mois [18]. Récemment, Teke et Citirik ont comparé deux séries de FC compliquées de décollement rétinien, l’une traitée par vitrectomie, laser et tamponnement par gaz, l’autre par vitrectomie seule. Les résultats anatomiques et fonctionnels ont été identiques dans les deux séries, mais la réapplication rétinienne a été significativement plus courte dans le groupe de patients ayant bénéficié d’un tamponnement par gaz (durée moyenne de réapplication : 20,9 ± 3,2 mois) par rapport au groupe traité par vitrectomie seule (durée moyenne de réapplication : 25,5 ± 4 mois) [22].
Enfin, Theodossiadis et al. ont proposé une indentation du pôle postérieur par une éponge sclérale pour limiter la migration du liquide intrarétinien et diminuer les tractions du vitré sur la rétine juxtapapillaire [23]. Les résultats anatomiques et visuels sont excellents, mais cette technique difficile nécessite un certain apprentissage.
eCas clinique 15-1 Femme âgée de 43 ans présentant une baisse de l’acuité visuelle de l’œil droit à 0,4.
[1] Georgalas I, Ladas I, Georgopoulos G, Petrou P. Optic disc pit : a review. Graefes Arch Clin Exp Ophthalmol 2011 ; 249 : 1113-22.
[2] Wiethe T. Ein Fall von angeborener Deformität der Sehnervenpapille. Arch Augenheilkd 1882 ; 11 : 14-9.
[3] Jain N, Johnson MW. Pathogenesis and treatment of maculopathy associated with cavitary optic disc anomalies. Am J Ophthalmol 2014 ; 158 : 423-35.
[4] Brodsky MC. Congenital optic disk anomalies. Surv Ophthalmol 1994 ; 39 : 89-112.
[5] Caputo G. Rapport de la Société Française d’Ophtalmologie. Issy-les-Moulineaux : Elsevier Masson 2011 ; 64 : 465-72.
[6] Ferry AP. Macular detachment associated with congenital pit of the optic nerve head. pathologic findings in two cases simulating malignant melanoma of the choroid. Arch Ophthalmol 1963 ; 70 : 346-57.
[7] Bonnet M. Serous macular detachment associated with optic nerve pits. Graefes Arch Clin Exp Ophthalmol 1991 ; 229 : 526-32.
[8] Gass JD. Serous detachment of the macula. Secondary to congenital pit of the optic nervehead. Am J Ophthalmol 1969 ; 67 : 821-41.
[9] Lincoff H, Lopez R, Kreissig I, et al. Retinoschisis associated with optic nerve pits. Arch Ophthalmol 1988 ; 106 : 61-7.
[10] Lincoff H, Kreissig I. Optical coherence tomography of pneumatic displacement of optic disc pit maculopathy. Br J Ophthalmol 1998 ; 82 : 367-72.
[11] Imamura Y, Zweifel SA, Fujiwara T, et al. High-resolution optical coherence tomography findings in optic pit maculopathy. Retina 2010 ; 30 : 1104-12.
[12] Roy R, Waanbah AD, Mathur G, et al. Optical coherence tomography characteristics in eyes with optic pit maculopathy. Retina 2013 ; 33 : 771-5.
[13] Annesley W, Brown G, Bolling J, et al. Treatment of retinal detachment with congenital optic pit by krypton laser photocoagulation. Graefes Arch Clin Exp Ophthalmol 1987 ; 225 : 311-4.
[14] Mustonen E, Varonen T. Congenital pit of the optic nerve head associated with serous detachment of the macula. Acta Ophthalmol 1972 ; 50 : 689-98.
[15] Akiyama H, Shimoda Y, Fukuchi M, et al. Intravitreal gas injection without vitrectomy for macular detachment associated with an optic disk pit. Retina 2014 ; 34 : 222-7.
[16] Sandali O, Barale PO, Bui Quoc E, et al. Résultats à long terme du traitement des fossettes colobomateuses associées à un décollement séreux de la macula : à propos de 20 cas. J Fr Ophtalmol 2011 ; 34 : 532-8.
[17] Yanyali A, Bonnet M. Traitement du décollement maculaire compliquant une fossette colobomateuse de la papille : résultats à long terme de l’association photocoagulation-gaz. J Fr Ophtalmol 1993 ; 16 : 523-31.
[18] Hirakata A, Inoue M, Hiraoka T, McCuen BW 2nd. Vitrectomy without laser treatment or gas tamponade for macular detachment associated with an optic disc pit. Ophthalmology 2012 ; 119 : 810-8.
[19] Shukla D, Kalliath J, Tandon M, Vijayakumar B. Vitrectomy for optic disk pit with macular schisis and outer retinal dehiscence. Retina 2012 ; 32 : 1337-42.
[20] Spaide RF, Fisher Y, Ober M, Stoller G. Surgical hypothesis : inner retinal fenestration as a treatment for optic disc pit maculopathy. Retina 2006 ; 26 : 89-91.
[21] Ooto S, Mittra RA, Ridley ME, Spaide RF. Vitrectomy with inner retinal fenestration for optic disc pit maculopathy. Ophthalmology 2014 ; 121 : 1727-33.
[22] Theodossiadis GP, Theodossiadis PG. Optical coherence tomography in optic disk pit maculopathy treated by the macular buckling procedure. Am J Ophthalmol 2001 ; 132 : 184-90.
[23] Teke MY, Citirik M. 23 Gauge vitrectomy, endolaser, and gas tamponade versus vitrectomy alone for serous macular detachment associated with optic disc pit. Am J Ophthalmol 2015 ; 160 : 779-85.
S.-Y. Cohen
➤ La dysversion papillaire est une anomalie congénitale correspondant à un axe oblique de la tête du nerf optique.
➤ L’atteinte papillaire s’accompagne d’une déformation de la partie inférieure du globe donnant un aspect d’« hémi-rétine de myope ».
➤ Les principales complications se situent au niveau du changement de courbure de ces deux hémi-rétines.
➤ Le décollement séreux rétinien parfois responsable de la baisse visuelle est fluctuant.
La dysversion papillaire est une anomalie congénitale relativement fréquente. Elle toucherait entre 1 et 3 % de la population générale [1, 2]. Elle correspond à un axe oblique de la tête du nerf optique associé à une antéroversion de la partie supérieure de la papille, d’où le terme de tilted disc dans la littérature anglo-saxonne. Différentes anomalies du fond d’œil peuvent être observées en association avec la dysversion : un croissant atrophique péripapillaire inférieur ou inféronasal ; des anomalies d’insertion des vaisseaux avec trajet initialement nasal des vaisseaux rétiniens desservant la partie temporale du fond d’œil ; un staphylome inférieur. L’anomalie est due à un retard de fermeture de la fente embryonnaire dans les tout premières semaines de la vie intra-utérine. Elle correspond à une forme mineure de colobome [2]. L’analyse en IRM tridimensionnelle a montré que l’anomalie principale est une déformation de la partie inférieure du globe située au-dessous du nerf optique avec un degré de myopie variable selon la position respective de la fovéa et de la protrusion inférieure [3].
La dysversion papillaire a plusieurs conséquences cliniques. L’acuité visuelle des patients est en règle inférieure à la normale. Ces patients présentent de plus un astigmatisme myopique, complexe. Les analyses du champ visuel peuvent mettre en évidence un scotome relatif pouvant ressembler à un scotome de type Bjerrum supérieur. Un amincissement de la couche des fibres optiques peut être observé autour de la papille.
Différentes complications ont été rapportées en association avec la dysversion papillaire : altérations pigmentaires en bande, situées à la jonction entre la rétine de courbure normale et le staphylome inférieur ou prenant un aspect en T [4, 5], néovascularisation choroïdienne (fig. 15-33) ou vasculopathie polypoïdale choroïdienne observée également dans la même zone de jonction [6, 7]. Plus rarement, des plis choriorétiniens [8] ou des anomalies de la jonction vitréorétinienne (membrane épirétinienne, aspect de trou lamellaire) ont été rapportés [9].
La complication spécifique nous intéressant dans le cadre de ce rapport est l’existence de décollements séreux rétiniens (fig. 15-34). Initialement, ces décollements séreux ont été décrits associés à des points de fuite évoquant fortement une choriorétinopathie séreuse centrale [10]. Ces décollements séreux surviennent dans la zone de jonction entre la courbure normale du globe et le staphylome inférieur. Ils ont été également observés à la jonction entre un staphylome postérieur et la rétine antérieure [11]. Il faut souligner la proximité de ces anomalies avec celles observées dans le syndrome de macula bombée. Il s’agit également d’une anomalie de courbure du globe oculaire se compliquant fréquemment d’un décollement séreux rétinien chronique [12].
Depuis l’utilisation courante de l’OCT maculaire, il est fréquent d’observer des zones de décollement séreux rétinien maculaire chez les patients atteints de dysversion papillaire. Cette anomalie a été observée dans 17 à 41 % des yeux présentant une dysversion.
L’angiographie à la fluorescéine ne montre pas toujours de point de fuite bien individualisé. L’aspect du fond d’œil peut alors être confondu soit avec une néovascularisation choroïdienne occulte, soit avec une choriorétinopathie séreuse centrale chronique d’autant que des altérations pigmentaires peuvent prendre un aspect en coulée gravitationnelle. Ces décollements séreux rétiniens ont fréquemment une évolution fluctuante avec des poussées ou des épisodes de réapplication de la rétine sensorielle.
L’origine exacte de ces décollements est inconnue. Cependant, il a été montré qu’à la partie inférieure de ces décollements pouvait être observé un amincissement choroïdien. Des turbulences hémodynamiques choroïdiennes pourraient donc être à l’origine de la formation de ces zones de soulèvement. Dans la macula bombée, un amincissement choroïdien périmaculaire est également observé. Les décollements séreux observés dans les deux affections pourraient donc avoir la même origine choroïdienne.
Des tentatives thérapeutiques ont été proposées. La photocoagulation au laser des points de fuite a été jugée efficace dans certains cas [10]. La thérapie photodynamique et les injections intravitréennes d’anti-VEGF ne semblent pas donner de résultats probants [13, 14]. Il faut souligner que la thérapie photodynamique présente également le risque d’altérer l’épithélium pigmentaire déjà probablement aminci chez ces patients. Ces décollements séreux rétiniens n’ont donc pas trouvé pour l’instant de solution thérapeutique acceptable.
[1] Vongphanit J, Mitchell P, Wang JJ. Population prevalence of tilted optic disks and the relationship of this sign to refractive error. Am J Ophthalmol 2002 ; 133 : 679-85.
[2] Apple DJ, Rabb MF, Walsh PM. Congenital anomalies of the optic disc. Surv Ophthalmol 1982 ; 27 : 3-41.
[3] Shinohara K, Moriyama M, Shimada N, et al. Analyses of shape of eyes and structure of optic nerves in eyes with tilted disc syndrome by swept-source optical coherence tomography and three-dimensional magnetic resonance imaging. Eye (Lond) 2013 ; 27 : 1233-41.
[4] Giuffrè G. Chorioretinal degenerative changes in the tilted disc syndrome. Int Ophthalmol 1991 ; 15 : 1-7.
[5] Cohen SY, Dubois L, Ayrault S, Quentel G. T-shaped pigmentary changes in tilted disk syndrome. Eur J Ophthalmol 2009 ; 19 : 876-9.
[6] Prost M, De Laey JJ. Choroidal neovascularization in tilted disc syndrome. Int Ophthalmol 1988 ; 12 : 131-5.
[7] Mauget-Faÿsse M, Cornut PL, Quaranta El-Maftouhi M, Leys A. Polypoidal choroidal vasculopathy in tilted disk syndrome and high myopia with staphyloma. Am J Ophthalmol 2006 ; 142 : 970-5.
[8] Cohen SY, Quentel G. Chorioretinal folds as a consequence of inferior staphyloma associated with tilted disc syndrome. Graefes Arch Clin Exp Ophthalmol 2006 ; 244 : 1536-8.
[9] Cohen SY, Dubois L, Nghiem-Buffet S, et al. Spectral domain optical coherence tomography analysis of macular changes in tilted disk syndrome. Retina 2013 ; 33 : 1338-45.
[10] Cohen SY, Quentel G, Guiberteau B, et al. Macular serous retinal detachment caused by subretinal leakage in tilted disc syndrome. Ophthalmology 1998 ; 105 : 1831-4.
[11] Leys AM, Cohen SY. Subretinal leakage in myopic eyes with a posterior staphyloma or tilted disk syndrome. Retina 2002 ; 22 : 659-65.
[12] Gaucher D, Erginay A, Lecleire-Collet A, et al. Dome-shaped macula in eyes with myopic posterior staphyloma. Am J Ophthalmol 2008 ; 145 : 909-14.
[13] Milani P, Pece A, Pierro L, et al. Bevacizumab for macular serous neuroretinal detachment in tilted disk syndrome. J Ophthalmol 2010 ; 2010 : 970580.
[14] Donati MC, Miele A, Abbruzzese G, et al. Treatment of macular serous neuroretinal detachment in tilted disk syndrome : report of 3 cases. Eur J Ophthalmol 2013 ; 23 : 267-70.
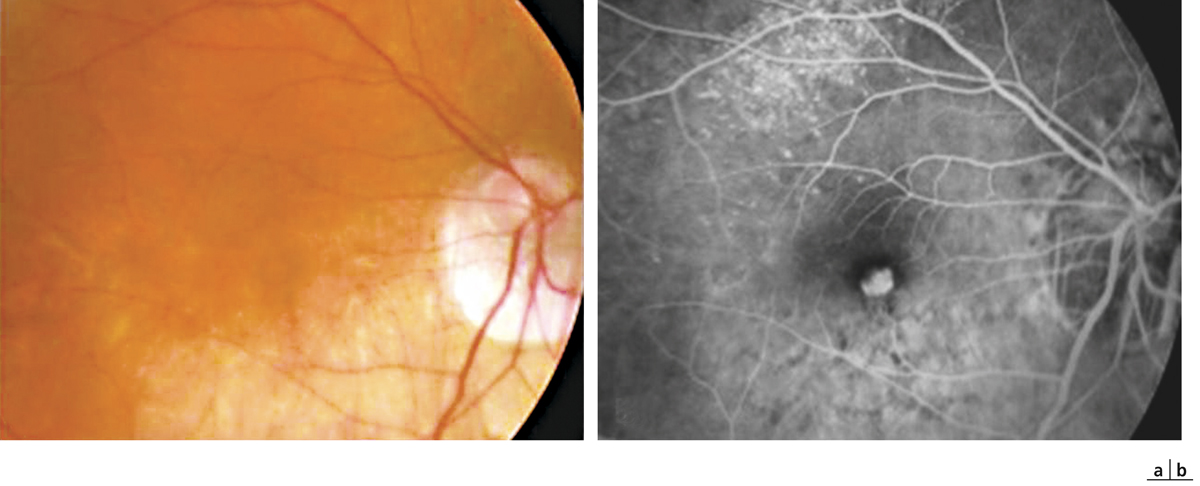
Fig. 15-33 Dysversion papillaire typique compliquée de néovascularisation choroïdienne.
a. Rétinographie en couleurs montrant une obliquité de la tête du nerf optique, un croissant « atrophique » inférieur, et une pâleur relative de la partie inférieure du fond d’œil, correspondant à l’aire du staphylome. b. Angiographie à la fluorescéine montrant une néovascularisation « visible » ou « classique », rétrofovéolaire, développée dans la zone de jonction entre le staphylome et la rétine de courbure normale.
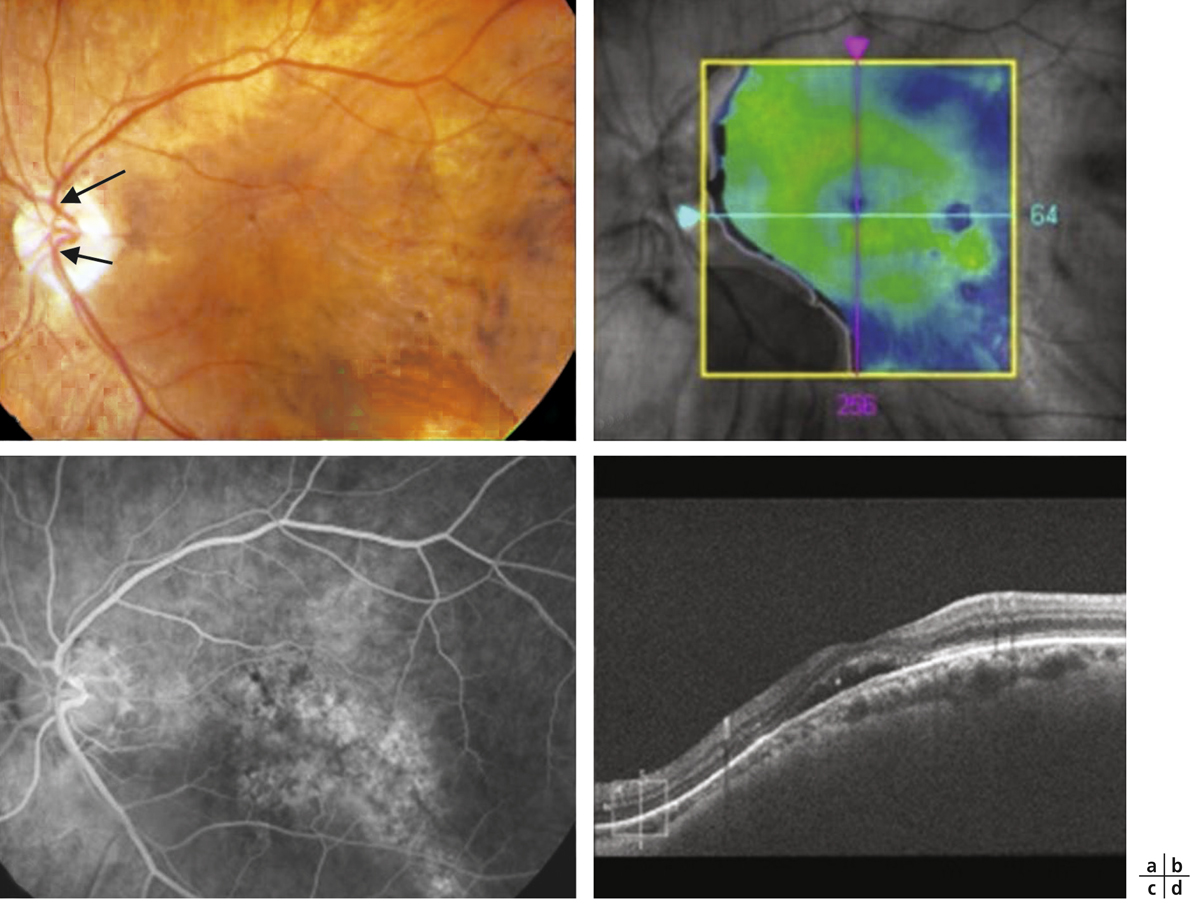
Fig. 15-34 Dysversion papillaire compliquée de décollement séreux rétinien.
a. Rétinographie en couleurs montrant une dysversion papillaire avec trajet anormal des vaisseaux rétiniens à la papille (départ à direction nasale, suivie d’un coude permettant une direction vers la partie temporale du fond d’œil, flèches). b. Cartographie incomplète de l’OCT très typique des anomalies de courbure du globe oculaire. c. Angiographie à la fluorescéine montrant des altérations pigmentaires dans l’aire de jonction entre le staphylome inférieur et la rétine de courbure normale. d. Coupe verticale de l’OCT montrant la présence d’un décollement séreux rétinien maculaire.
A. Bron, C. Vignal, C. Creuzot-Garcher
➤ Les œdèmes maculaires microkystiques (OMM) constituent une nouvelle entité clinique mise en évidence grâce aux progrès de l’OCT. Ces lésions apparaissent comme des structures kystiques hyporéflectives périmaculaires. Ces faux œdèmes ne sont pas l’apanage des neuropathies optiques. Leur signification et leurs conséquences cliniques demandent à être précisées dans des séries plus nombreuses et longitudinales.
➤ La maculopathie par hypotonie survient dans des situations prévisibles (trabéculectomie, cyclodialyse traumatique) et demande un examen soigneux pour bien la dépister et suivre son évolution. Son diagnostic précoce est indispensable pour éviter de pérenniser cette situation source de baisse visuelle.
Des aspects trompeurs d’œdème maculaire peuvent se rencontrer dans plusieurs situations cliniques. Ici nous décrirons deux tableaux cliniques qui peuvent orienter vers un œdème maculaire, les œdèmes maculaires microkystiques (OMM) et les maculopathies par hypotonie.
Quel que soit leur mécanisme, les neuropathies optiques (NO) ont comme conséquence une diminution du nombre des cellules ganglionnaires. L’atrophie optique (AO) est ainsi l’évolution terminale commune des atteintes du nerf optique, qu’elles soient glaucomateuses, vasculaires, inflammatoires – liées ou non à une sclérose en plaques –, héréditaires, toxiques ou compressives. L’examen clinique permet de visualiser la pâleur du disque optique (fig. 15-35a) et le champ visuel de quantifier l’altération fonctionnelle. L’examen OCT avec le mode retinal nerve fiber layer (RNFL) quantifie la perte en fibres et permet de suivre l’évolution de cette dernière en mesurant l’épaisseur globale et, dans chaque quadrant, de la couche des fibres ganglionnaires péripapillaires et, plus récemment, du complexe ganglionnaire (fig. 15-35b). Cette perte en fibres ganglionnaires induit également classiquement une diminution de l’épaisseur maculaire mesurée par le volume maculaire en OCT.
L’utilisation du SD-OCT à haute résolution a permis de segmenter et de mieux visualiser les différentes couches de la rétine. L’usage généralisé de l’OCT dans les pathologies maculaires et celles du nerf optique a permis de mettre en évidence ce qu’il est convenu d’appeler aujourd’hui les œdèmes maculaires microkystiques (OMM). Pour être plus précis, car les lésions observées ne possèdent pas de coque épithéliale bien limitée, il paraît plus juste d’employer le terme de cystoïde à celui de kystique [1]. La terminologie ne cesse d’évoluer et certains auteurs ont évoqué le terme de maculopathie rétrograde impliquant un dysfonctionnement axonal des cellules ganglionnaires de la rétine [2]. Ce terme est cependant erroné et restrictif car l’on sait maintenant que ces lésions ne sont pas spécifiques des neuropathies optiques mais de plusieurs pathologies rétiniennes [3].
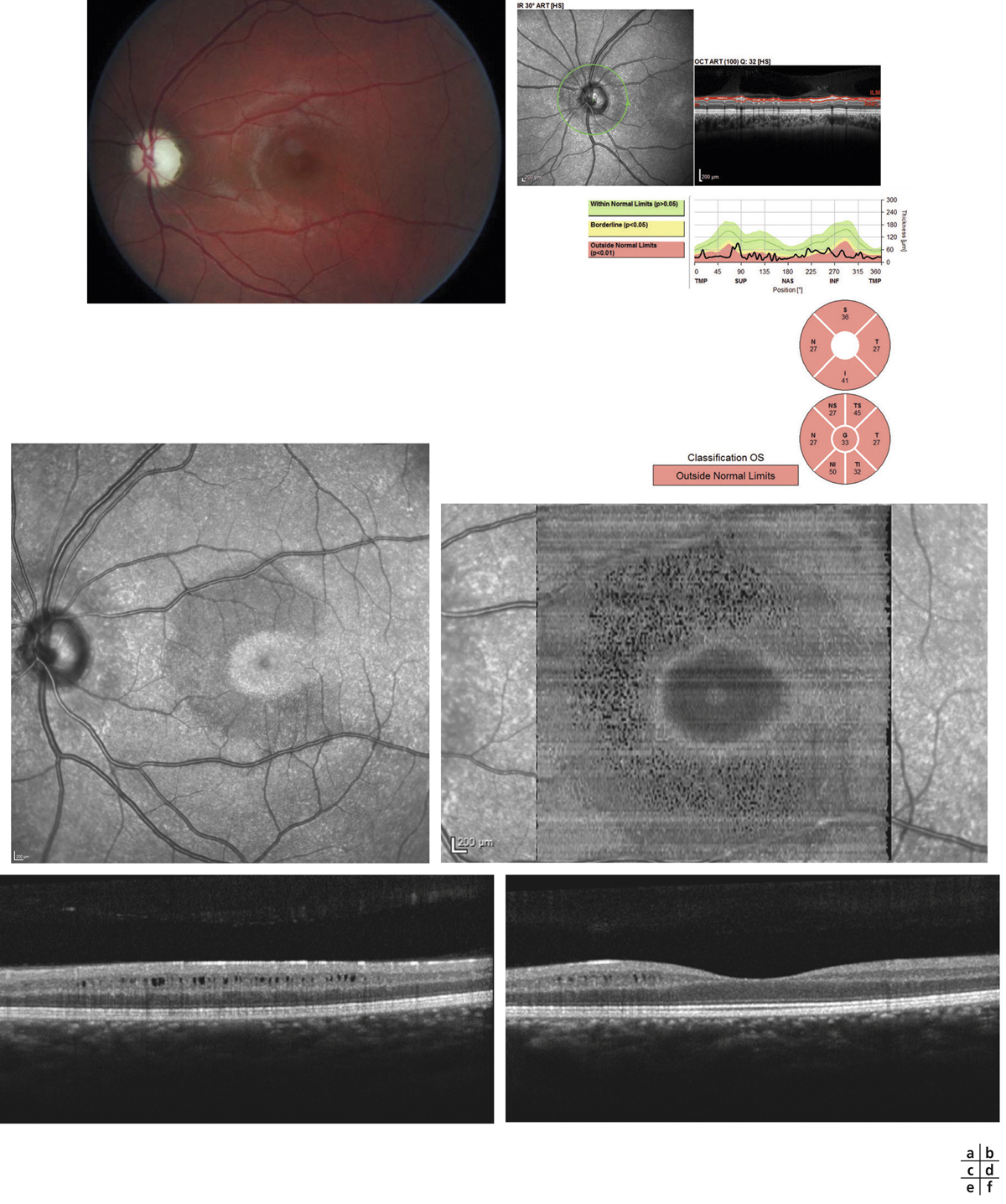
Fig. 15-35 Patiente de 25 ans, antécédent de névrite optique gauche sévère avec acuité inférieure à 1/20.
a. Cliché en couleurs de l’œil gauche montrant la pâleur papillaire. b. OCT RNFL (Spectralis®, Heidelberg, Allemagne) : importante perte en fibres dans tous les quadrants. c. OCT Spectralis®, cliché infrarouge : image en cocarde hyporéflective correspondant aux microkystes. d. OCT Spectralis®, cliché « en face » permettant de visualiser les kystes. e, f. Coupes longitudinales en OCT Spectralis® (passant en supérieur (e) et au niveau maculaire (f) visualisant les kystes au niveau de la couche nucléaire interne.
(Source : collection de V. Vasseur.)
Identifiées initialement en 2012 par Gelfand et al., ces lésions apparaissent comme des structures kystiques hyporéflectives situées autour de la macula, au niveau de la couche nucléaire interne [4]. Elles ont été initialement identifiées lors d’atrophie optique chez des patients atteints de sclérose en plaques (SEP) avec des antécédents de névrites optiques. Ces îlots cystoïdes, bien délimités, doivent être présents sur au moins deux coupes d’OCT. Ces lésions hyporéflectives siègent dans la région périmaculaire et sont localisées dans la couche nucléaire interne en nasal ou en temporal de la fovéa [3].
Leur délai d’apparition et leur fréquence par rapport à la NO sont mal définis ; cependant l’OMM semble plus fréquent en cas d’AO sévère et sa présence reflète en règle une fonction visuelle médiocre. Leur topographie dans la zone périmaculaire est variable et parfois évolutive au cours du temps ; l’OMM prend le plus souvent la forme d’un croissant ou d’une cocarde plus ou moins complète et est mieux visualisé sur les clichés infrarouges (fig. 15-35c) sous la forme d’une zone sombre hyporéflective [5], ainsi que sur les images d’OCT « en face » (fig. 15-35d).
En cas d’OMM, il existe une augmentation relative de l’épaisseur maculaire par rapport au côté sain. Sur les coupes linéaires d’OCT, les microkystes sont des structures rondes ou ovalaires, hyporéflectives situées dans la couche nucléaire interne (fig. 15-35e et f). Leur taille est variable allant de 20–30 à 70–90 µ et, à la différence d’un œdème maculaire classique, ils n’entraînent pas de diffusion de colorant à l’angiographie à la fluorescéine lorsque celle-ci est pratiquée. Celle-ci n’est toutefois pas nécessaire, car il ne s’agit pas d’un œdème rétinien [5].
Depuis 2012, la littérature s’est enrichie de nouveaux cas d’OMM et à la fin 2013 environ 178 yeux de 127 patients avaient été rapportés dans la littérature [3]. Une série monocentrique de 142 yeux de 133 patients avec un OMM a rapporté en 2014 une prévalence de 10 % d’OMM sur une série rétrospective de 1368 patients présentant une neuropathie optique [3]. Aucun des sujets contrôles ne présentait d’OMM. C’est dire si ce qui était au départ une curiosité, est probablement beaucoup plus fréquent que l’on pouvait le croire lors des premières descriptions.
Les deux sexes semblent touchés également et les tranches d’âge plus concernées sont les jeunes pour les uns [2], alors que dans la série la plus importante l’âge moyen est voisin de 73 ans [3].
La cause du développement de ces microkystes est mal connue. Des vacuoles identiques à ces kystes ont été décrites chez des singes présentant une atrophie optique préexistante ; leur présence lors de l’évolution des AO suggère que la dégénérescence rétrograde transsynaptique conduit à une dégénérescence au niveau de la nucléaire interne avec formation de kystes. Ces derniers reflètent probablement la dégénérescence des cellules de Müller associée à la perte des fibres optiques après une NO, probablement par un mécanisme impliquant le flux d’eau (via les aquaporines) et le métabolisme du glutamate. Ils confirment les rapports étroits entre les structures de la macula et du nerf optique (fig. 15-36).
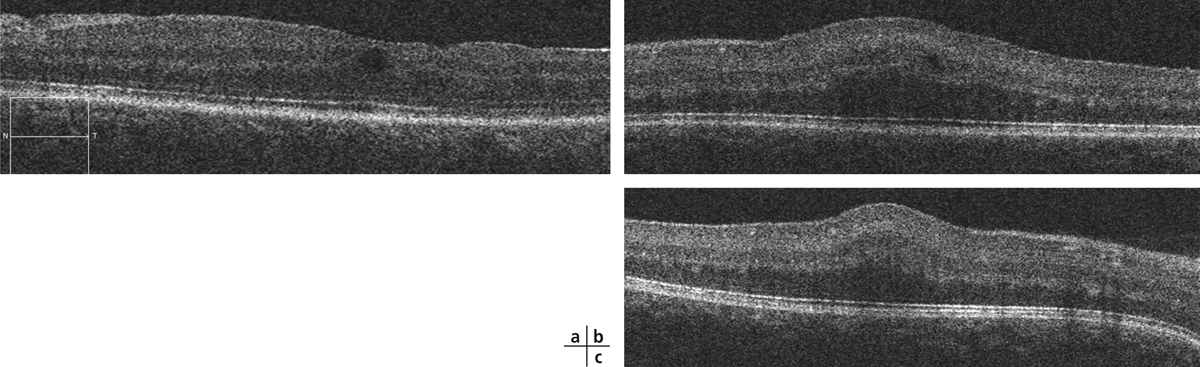
Fig. 15-36 Patients présentant un œdème maculaire microkystique.
a. Chez un patient présentant une neuropathie toxique au méthanol après absorption d’alcool frelaté au cours d’un voyage en Asie. b, c. Chez une enfant de 16 ans nanophtalme et porteuse d’une maladie de Best (b). Cette association permet d’évoquer la participation de gènes codant pour des protéines qui assurent l’étanchéité intercellulaire, les frizzled proteins (protéines de jonction). Une anomalie de ces jonctions intercellulaires conduit à une fuite de matériel cellulaire ou de fluide qui se matérialise par ces dépôts dans la rétine externe de la maladie de Best, une effusion choroïdienne et l’existence des OMM. Noter la perte du profil fovéolaire habituelle chez les nanophtalmes. Même patiente 1 an plus tard (c) : la lésion cystoïde a disparu.
Si les premiers cas concernaient des patients porteurs de SEP avec des NO inflammatoires [4], on sait maintenant qu’on peut les trouver dans de multiples étiologies (encadré 15-1
• Tous types de neuropathie optique : glaucome ; neuropathies toxique, compressive ou ischémique
• Dégénérescences maculaires liées à l’âge
• Membranes épirétiniennes, après chirurgie maculaire
• Rétinopathie diabétique
• Occlusions vasculaires
• Uvéites
• Choroïdite séreuse centrale idiopathique
• Rétinopathies pigmentaires [2, 6, 7]
• Télangiectasies maculaires idiopathiques du groupe 2A
• Rétinopathie au tamoxifène [3, 8]
• Les pathologies du nerf optique
). Les avis restent partagés concernant leur évolution : elles semblent s’aggraver pour Abegg, alors qu’au contraire elles sont transitoires dans 84 % des cas pour Burggraaff [2, 3]. Cependant ces avis partent de pathologies associées très différentes : des NO dans le premier cas et des pathologies variées dans le second.
Le pronostic est également diversement apprécié : pour les uns, les OMM s’accompagnent d’une baisse d’acuité visuelle [2], alors que l’acuité visuelle n’est pas corrélée à l’existence des OMM pour les autres [3]. Les anomalies associées sont principalement une modification de l’épaisseur de la nucléaire interne qui, suivant les séries, est soit augmentée, soit diminuée [3, 9].
Décrites pour la première fois en 1954 [10], les maculopathies par hypotonie peuvent être confondues avec un œdème maculaire, mais dans un certain nombre de cas elles s’accompagnent d’un authentique œdème maculaire, le plus souvent cystoïde [11].
La situation clinique est celle d’une hypotonie oculaire, généralement inférieure à 6 mmHg, associée à des anomalies du fond d’œil à type de plis choriorétiniens, d’œdème papillaire et de tortuosité des vaisseaux. La baisse d’acuité visuelle de loin est plus ou moins constante avec une modification de la réfraction vers l’hypermétropie. L’acuité visuelle de près est plus volontiers atteinte. À la lampe à fente, on pourra retrouver suivant l’étiologie une fuite au niveau de la bulle de la filtration ou bien une bulle de filtration excessive après trabéculectomie. En cas de traumatisme oculaire, la gonioscopie pourra révéler des modifications angulaires à type de cyclodialyse. C’est surtout l’examen du fond d’œil qui permettra de porter le diagnostic de maculopathie par hypotonie. La papille est œdémateuse avec des vaisseaux tortueux et l’on note un bombement antérieur de la macula avec des plis maculaires interpapillomaculaires et temporaux (fig. 15-37).
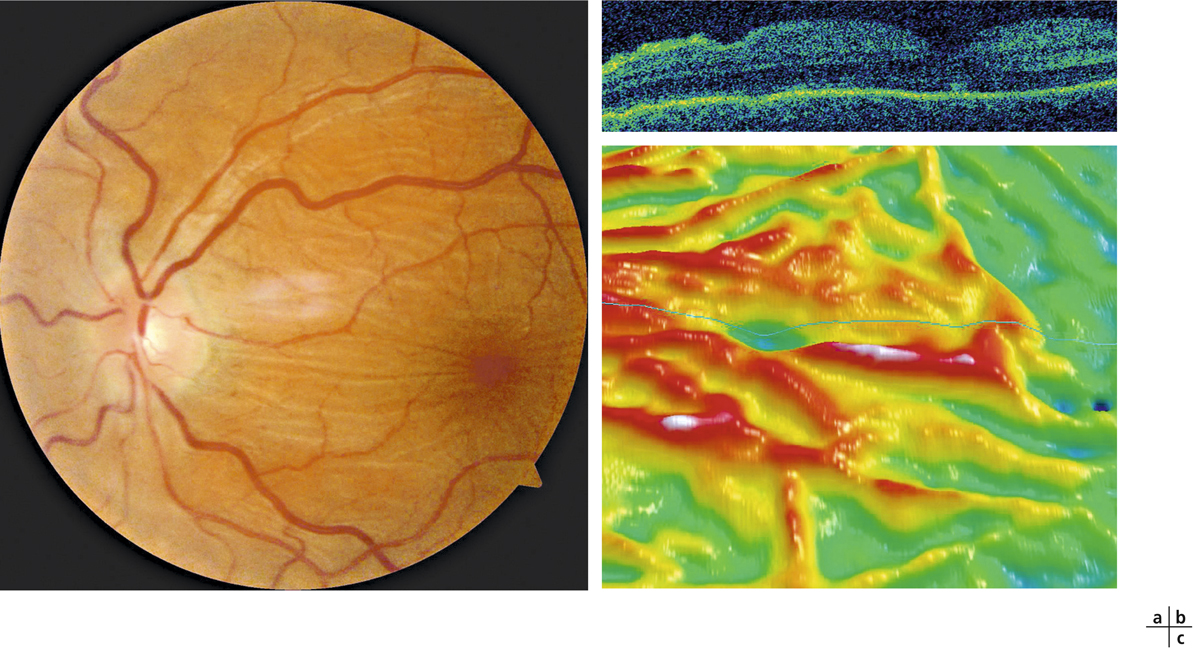
Fig. 15-37 Maculopathie par hypotonie après chirurgie de trabéculectomie avec mitomycine C chez une jeune femme myope porteuse d’un glaucome pigmentaire.
a. L’œdème papillaire, la tortuosité des vaisseaux et les plis maculaires sont bien visibles. b. OCT : les plis maculaires sont bien mis en évidence. c. Cartographie rétinienne 3D montrant l’épaississement de la rétine et le pseudo-œdème maculaire.
Les maculopathies par hypotonie se rencontrent principalement dans deux circonstances : après chirurgie de glaucome et après traumatisme oculaire. L’usage des antimétabolites comme la mitomycine C dans la chirurgie des glaucomes est particulièrement pourvoyeur de cette complication. Son effet toxique sur le corps ciliaire est redouté, notamment pour les sclères fines qui favorisent sa diffusion. Il est habituel d’avoir une hypotonie les jours suivants une sclérectomie profonde. Cette hypotonie, qui oscille entre 0 et 5 mmHg, est très particulière puisque la chambre antérieure est ici bien formée et qu’il n’y a pas de retentissement au niveau du segment postérieur. Dans les séries un peu anciennes, la maculopathie par hypotonie pouvait atteindre 20 % des cas opérés de trabéculectomie [12, 13]. Plus récemment sur un groupe de 428 yeux opérés de trabéculectomie en Angleterre, cette complication a été retrouvée précocement dans 2,6 % des cas et plus tardivement dans 7,2 % des cas [14].
Le diagnostic clinique est précisé par l’OCT qui définit l’étendue des plis rétiniens et l’éventuel œdème maculaire associé (fig. 15-37). Un OCT de segment antérieur permettra de mieux mettre en évidence une cyclodialyse. L’angiographie fluorescéinique n’est pas nécessaire de même que l’échographie.
Tous les yeux « mous » ne présentent pas de maculopathie par hypotonie. Certains auteurs ont retrouvé l’âge jeune, le sexe masculin et la myopie comme facteurs de risque [15, 16]. La rigidité sclérale semble entrer en jeu avec une formation de plis et un bombement antérieur de la sclère située derrière la macula comme ce qui a pu être évoqué pour la macula bombée chez le myope fort [11].
Le traitement dépend de l’étiologie et repose sur un examen soigneux. Après chirurgie de trabéculectomie, si une fuite au niveau conjonctival est détectée, il faudra réparer la conjonctive par différents moyens qui vont de la colle à la reprise du surjet. En cas de sutures trop lâches de la trappe de filtration, il faudra reprendre ces sutures et plus volontiers par le moyen de sutures transconjonctivales dites de compression [17].
Après traumatisme (fig. 15-38), la réinsertion du corps ciliaire à la sclère pourra s’avérer nécessaire mais ce n’est pas un geste facile. Les corticoïdes par voie topique sont souvent prescrits pour tenter de remonter la pression intra-oculaire ; ils peuvent certes aider, mais leur efficacité est inconstante.
Fig. 15-38 Maculopathie par hypotonie après traumatisme oculaire violent avec cyclodialyse modérée.
[1] Sigler EJ. Microcysts in the inner nuclear layer, a nonspecific SD-OCT sign of cystoid macular edema. Invest Ophthalmol Vis Sci 2014 ; 55 : 3282-4.
[2] Abegg M, Dysli M, Wolf S, et al. Microcystic macular edema : retrograde maculopathy caused by optic neuropathy. Ophthalmology 2014 ; 121 : 142-9.
[3] Burggraaff MC, Trieu J, de Vries-Knoppert WA, et al. The clinical spectrum of microcystic macular edema. Invest Ophthalmol Vis Sci 2014 ; 55 : 952-61.
[4] Gelfand JM, Nolan R, Schwartz DM, et al. Microcystic macular oedema in multiple sclerosis is associated with disease severity. Brain 2012 ; 135 : 1786-93.
[5] Wolff B, Basdekidou C, Vasseur V, et al. Retinal inner nuclear layer microcystic changes in optic nerve atrophy : a novel spectral-domain OCT finding. Retina 2013 ; 33 : 2133-8.
[6] Balk LJ, Killestein J, Polman CH, et al. Microcystic macular oedema confirmed, but not specific for multiple sclerosis. Brain 2012 ; 135 : e226 ; author reply e227.
[7] Wolff B, Azar G, Vasseur V, et al. Microcystic changes in the retinal internal nuclear layer associated with optic atrophy : a prospective study. J Ophthalmol 2014 ; 2014 : 395189.
[8] Cohen SY, Dubois L, Nghiem-Buffet S, et al. Retinal pseudocysts in age-related geographic atrophy. Am J Ophthalmol 2010 ; 150 : 211-217.e1.
[9] Kaufhold F, Zimmermann H, Schneider E, et al. Optic neuritis is associated with inner nuclear layer thickening and microcystic macular edema independently of multiple sclerosis. PLoS One 2013 ; 8 : e71145.
[10] Dellaporta A. Creasing of retina in hypotonia. Klin Monbl Augenheilkd Augenarztl Fortbild 1954 ; 125 : 672-8.
[11] Costa VP, Arcieri ES. Hypotony maculopathy. Acta Ophthalmol Scand 2007 ; 85 : 586-97.
[12] Kitazawa Y, Suemori-Matsushita H, Yamamoto T, Kawase K. Low-dose and high-dose mitomycin trabeculectomy as an initial surgery in primary open-angle glaucoma. Ophthalmology 1993 ; 100 : 1624-8.
[13] Megevand GS, Salmon JF, Scholtz RP, Murray AD. The effect of reducing the exposure time of mitomycin C in glaucoma filtering surgery. Ophthalmology 1995 ; 102 : 84-90.
[14] Kirwan JF, Lockwood AJ, Shah P, et al. Trabeculectomy in the 21st century : a multicenter analysis. Ophthalmology 2013 ; 120 : 2532-9.
[15] Jampel HD, Pasquale LR, Dibernardo C. Hypotony maculopathy following trabeculectomy with mitomycin C. Arch Ophthalmol 1992 ; 110 : 1049-50.
[16] Stamper RL, McMenemy MG, Lieberman MF. Hypotonous maculopathy after trabeculectomy with subconjunctival 5-fluorouracil. Am J Ophthalmol 1992 ; 114 : 544-53.
[17] Eha J, Hoffmann EM, Wahl J, Pfeiffer N. Flap suture--a simple technique for the revision of hypotony maculopathy following trabeculectomy with mitomycin C. Graefes Arch Clin Exp Ophthalmol 2008 ; 246 : 869-74.