Réhabilitation de la perception
C. Milleret
Dans le monde, on compte actuellement des dizaines de millions de personnes souffrant de pathologies rétiniennes et devenues aveugles. Leur perception du monde extérieur s’en trouve inévitablement altérée. Toutefois, au moins dans certains cas, ces personnes aveugles compensent leur handicap par les autres modalités sensorielles telles que l’audition, le toucher et le goût qui deviennent plus performantes que la normale. C’est la compensation transmodale. À cet effet, des réorganisations neuronales importantes s’opèrent dans les structures centrales normalement dédiées à ces « modalités de substitution », mais également dans le cortex visuel qui se met aussi à intégrer ces modalités sensorielles autres que la vision.
Après un bref historique général, nous allons voir ici plus particulièrement comment l’aveugle total précoce (ATP) compense sa perte de vision via l’audition. Il s’agit de la compensation transmodale la plus connue. La cécité totale a été choisie car la cécité partielle est mal compensée via un processus transmodal, sans doute du fait d’un conflit entre la vision résiduelle et les autres modalités sensorielles [1]. Le choix de l’aveugle précoce (jusqu’à environ 6 ans chez l’enfant) a par ailleurs été dicté par le fait que la plasticité de son cerveau et, donc, ses capacités d’adaptation sont bien plus grandes que celles de l’aveugle tardif, même si ce dernier peut encore s’adapter au handicap visuel [2].
La vision est une modalité sensorielle importante, si ce n’est la plus importante, puisqu’elle joue un rôle majeur dans le cadre de nos interactions avec l’environnement et est étroitement liée à l’action. Aussi, au fil des siècles, nombreux sont ceux qui se sont interrogés sur les conséquences de la cécité, à commencer par les philosophes. Dès le xviiie siècle, Diderot a émis l’hypothèse que les aveugles ont la capacité de compenser leur handicap en utilisant les autres sens tels que l’audition et le toucher. Dans sa Lettre sur les aveugles à l’usage de ceux qui voient publiée en 1749, il rapporte le fameux cas d’un mathématicien capable de différencier des pièces de monnaie par le simple toucher. Bien évidemment, d’autres pensaient au contraire que, sans vision, les autres modalités sensorielles ne pouvaient se développer normalement, la vision tant en quelque sorte la « référence » nécessaire à leur bon développement. Comme nous allons le voir ci-dessous, une approche plus scientifique de la question a donné raison à Diderot, mais seulement à la fin du xxe siècle !
Les premières études « scientifiques » ayant eu pour objectif de quantifier objectivement les capacités adaptatives transmodales de l’ATP face à son handicap ont été comportementales et psychophysiques, menées chez l’homme et l’animal. Elles ont eu pour objectif d’aborder le problème de la localisation spatiale via l’audition et celui de la discrimination de sons divers chez l’ATP.
Ces premières études ont révélé que l’ATP peut avoir une meilleure habileté à localiser les sons que le sujet voyant, quelle que soit la position des sons dans l’espace, même avec une seule oreille [1, 3, 4]. Mais ce n’est pas systématique d’un sujet à l’autre [1]. L’ATP peut aussi être meilleur que le sujet voyant pour évaluer la distance qui le sépare d’une source sonore [2, 5].
Concernant la discrimination des sons, il a été démontré que l’ATP pouvait être aussi meilleur pour identifier les changements de tonalité, même lorsqu’ils sont 10 fois plus rapides que ceux percevables par le sujet contrôle [6]. Il peut également être meilleur pour discriminer des voix et identifier une voix « cible », même dans une ambiance bruitée [7, 8]. L’ATP peut encore avoir la faculté de mieux discriminer des syllabes [9]. De telles facultés font de certains d’entre eux des musiciens d’exception, avec une oreille absolue.
Globalement, la cécité totale précoce peut être compensée par la perception auditive. La question est maintenant de savoir comment.
Ce sont des études fonctionnelles et anatomiques, conduites chez l’animal et chez l’homme, qui ont permis de mieux comprendre comment l’ATP peut posséder des supracapacités perceptives auditives. Il s’est avéré que son système auditif et son système visuel participent à la genèse de ces supraperformances via des réorganisations neuronales diverses.
Peu de données sont aujourd’hui disponibles concernant les réorganisations neuronales qui s’opèrent spécifiquement au niveau du système auditif et qui peuvent rendre compte des supraperformances auditives observées chez l’ATP. Toutefois, le rôle de ce système est réel et avéré. L’audition au niveau de l’oreille elle-même reste « normale ». Mais les structures centrales auditives voient leurs caractéristiques fonctionnelles changer après cécité. Ainsi, les neurones des aires corticales auditives (Fig. 9-1a, en vert) deviennent plus précis que la normale pour coder la localisation spatiale des sons [10,11]. Ces mêmes aires deviennent aussi plus grandes chez l’ATP que chez le sujet voyant [12, 13]. La cécité a par ailleurs pour conséquences une réduction de la latence et une augmentation de la robustesse des réponses auditives dans le cortex auditif chez l’ATP comparativement au sujet voyant [13,14].
Contrairement au système auditif, les réorganisations qui s’opèrent au niveau du système visuel lors des processus de compensation transmodale chez l’ATP sont plutôt bien connues. En effet, elles ont fait, et font encore actuellement, l’objet de nombreuses études tant chez l’animal que chez l’homme. Pour la plupart, ces études se sont toutefois concentrées sur le cortex. En effet, son implication a surpris, intrigue et est porteuse d’espoir pour le futur (Fig. 9-1a, en bleu).
C’est en 1988 et à l’aide d’une technique d’imagerie cérébrale, la tomographie à émission de positrons (TEP), que fut découverte pour la première fois l’activation du cortex visuel primaire (et des aires corticales adjacentes V2 et V3) lors d’une tâche de localisation spatiale de sons chez l’ATP humain [15]. Ce résultat important fut d’abord contesté car inattendu (!). Mais il fut finalement accepté car confirmé à plusieurs reprises. À cette occasion, il a en outre été montré que les performances de localisation des sons de l’ATP sont d’autant plus élevées que l’activation corticale est importante [16]. En revanche, elles deviennent nulles si l’on applique une stimulation directe de la région occipitale du cerveau par TMS (transcranial magnetic stimulation ou stimulation magnétique transcrânienne) pendant cette même tâche [17]. Plusieurs autres études menées chez l’homme et chez l’animal ont par ailleurs montré la réactivité neuronale effective du cortex visuel primaire à des stimulations auditives de diverses fréquences [14, 18, 19].
Le traitement de l’information visuelle chez le sujet voyant ne se limite pas aux seules aires visuelles corticales V1, V2 et V3 (voir chap. 2-1). Ce traitement s’effectue en effet au niveau de nombreuses autres aires visuelles corticales, dites supérieures. Alimentées par les aires V1, V2 et V3, elles forment en particulier la voie dorsale qui répond à la question « Où est-ce ? » et « Comment ? », et la voie ventrale qui répond à la question « Qu’est-ce que c’est ? » (Fig. 9-1a). Logiquement, la question de savoir si ces deux voies conservent les mêmes fonctions chez l’ATP s’est posée et la réponse est assurément positive [16, 20-22]. Globalement, cela suggère très fortement que l’ATP continue à « voir » avec son système visuel mais via l’audition, d’autant plus qu’il a été montré récemment que la connectivité fonctionnelle du système visuel chez l’ATP préserve la rétinotopie, c’est-à-dire une représentation ordonnée de l’espace au niveau de chacune des structures qui le constitue [23].

Fig. 9-1 Système visuel, système auditif et interactions chez le sujet voyant et le sujet aveugle.
a. Aires visuelles (en bleu) et aires auditives (en vert) au niveau du cortex cérébral du sujet voyant et de l’aveugle total précoce. Dans les eux cas, les aires visuelles primaires reçoivent des afférences issues des aires auditives (flèche verte). Mais elles sont inhibitrices chez le sujet voyant alors qu’elles sont excitatrices chez l’aveugle précoce. b. Interactions entre le système auditif et le système visuel. En vert, voie auditive primaire commune au sujet voyant et au sujet aveugle. En bleu, voies empruntées par les informations auditives pour parvenir au cortex visuel. 1. Voie commune aux sujets voyants et aveugles. Son activité est accrue chez l’aveugle via une levée d’inhibition (voir texte). 2. Voie existant stricteent chez le sujet aveugle. Au lieu de recevoir les afférences visuelles rétiniennes, le corps genouillé latéral dorsal reçoit ses afférences via le coliculus inférieur, c’est-à-dire une structure mésencéphalique typiquement auditive. AAS : aires auditives supérieures ; D : droite ; G : gauche.
Tous les mécanismes susceptibles d’être impliqués dans la compensation transmodale qui se développe chez l’ATP via l’audition ne seront pas évoqués ici, d’autant plus que la plupart d’entre eux sont encore mal connus. Seront seulement abordés ceux qui, à ce jour, sont bien établis ou semblent les plus importants.
Tout d’abord, il convient d’évoquer le fait que la mise en place de l’organisation générale du système visuel des mammifères, et donc la spécialisation fonctionnelle sous-jacente des structures qui le composent, est gérée majoritairement par des facteurs génétiques, sans aucune expérience visuelle, y compris chez l’homme [24]. Aussi ne doit-on pas être surpris par le fait que l’ATP va continuer à utiliser ce système pour « voir », même s’il fonctionne dorénavant avec des informations auditives.
Chez le voyant, la maturation du système visuel est toutefois très dépendante de l’expérience visuelle postnatale, en particulier pendant la période sensible (ou période critique) [25]. Si la vision est totalement absente depuis la naissance ou perdue pendant la période sensible après la naissance, le système visuel non seulement va rester à un stade immature, mais il va aussi se détériorer avec le temps, tant d’un point de vue anatomique que fonctionnel, avec en particulier une diminution drastique du nombre de ses connexions. Mais, comme décrit ci-dessus, dans le même temps, les autres modalités sensorielles telles que l’audition vont se substituer à la vision pour compenser ce handicap. Aussi les caractéristiques anatomofonctionnelles du système visuel vont-elles bien évidemment s’adapter pour remplir ce rôle. La question est maintenant de savoir comment. À cet effet, plusieurs mécanismes peuvent être évoqués :
les réponses auditives déjà présentes dans le cortex visuel du sujet voyant sont accrues après cécité totale précoce. Chez le sujet voyant, le cortex visuel (comme tous les autres cortex primaires d’ailleurs) n’est en effet pas unimodal : en plus de la vision, il reçoit des informations concernant toutes les autres modalités via des connexions cortico-corticales (intracorticales et interhémisphériques), mais celles-ci sont inhibées. Chez l’ATP, en revanche, ces inhibitions sont levées et ces connexions deviennent excitatrices [26] (Fig. 9-1a, flèche verte ; Fig. 9-1b, voie 1) ;
de nouvelles réponses auditives vont s’ajouter aux précédentes dans le cortex visuel de l’ATP du fait de la mise en place de nouvelles voies anatomiques. À cet effet, même si tous les auteurs ne sont pas d’accord et qu’il semble exister des différences interspécifiques, ces nouvelles informations auditives semblent gagner le cortex visuel primaire chez l’ATP via une voie sous-corticale principale : le colliculus inférieur >>> corps genouillé latéral dorsal (CGLd) >>> aires V1 et sans doute V2 [27] (Fig. 9-1b, voie 2) ;
l’enrichissement et l’entraînement semblent déterminer le degré de compensation transmodale chez l’ATP, comme attendu,substituplus ceux-ci sont importants et associés à un bon état attentionnel, meilleure est la compensation [19].
Pour une modalité sensorielle donnée, on a déjà mentionné que le degré de compensation transmodale via l’audition varie d’un sujet à l’autre, de très élevé à nul [1 et données personnelles]. Les raisons d’une telle hétérogénéité sont inconnues. L’âge de survenue de la cécité (avant la naissance, pendant la période sensible, etc.) ne semble pas entrer en ligne de compte. Aussi doit-on envisager que certains ATP utilisent plus volontiers une modalité qu’une autre, par exemple plus le toucher que l’audition. Mais les raisons d’un tel état de fait restent à identifier.
Lorsque c’est possible, et sur la base de ce qui est écrit ci-dessus, il faut essayer de redonner la vision à un ATP qui n’a jamais vu ou qui a bénéficié d’une expérience visuelle postnatale précoce. L’intervention doit toutefois avoir lieu aussitôt que possible, c’est-à-dire pendant la période sensible. La vision qu’on donne (ou redonne) au sujet doit de plus être de bonne qualité car structurante. En revanche, il convient de s’abstenir dans le cas d’un ATP adulte qui n’a jamais vu. En effet, dans ce cas, le système visuel, même en place, s’est adapté à compenser le handicap via les autres modalités sensorielles, dont l’audition, en établissant de nouveaux réseaux neuronaux tout à fait fonctionnels, impossibles à défaire et qui dorénavant sous-tendent diverses fonctions cognitives fondamentales, telle par exemple la représentation de l’espace indispensable à la navigation. Redonner la vision dans ce dernier cas conduira inévitablement à un conflit perceptif ingérable pour l’ATP, qui perdra alors tout repère, comme l’attestent d’ailleurs quelques cas déjà bien connus.
Compte tenu de ce qui précède, la question de savoir comment aider les ATP adultes en améliorant leurs performances perceptives s’est névitablement posée. Les ingénieurs et scientifiques ont abordé cette question dès les années 1970 en développant différents systèmes de substitution sensoriels ayant pour principe de transformer la scène visuelle captée par une caméra placée sur la tête du sujet non voyant en signaux somesthésiques ou auditifs [28, 29]. Sans entrer dans les détails, on cherche aujourd’hui à optimiser un tel concept (voir les travaux d’Amir Amedi en général) [30, 31]. Il est évident qu’une complète compréhension des processus de compensation transmodale résumés ci-dessus permettrait d’optimiser ces systèmes d’aide.
[1] Lessard N, Paré M, Lepore F, et al. Early-blind human subjects localize sound sources better than sighted subjects. Nature 1998 ; 395(6699) : 278-80.
[2] Voss P, Lassonde M, Gougoux F, et al. Early-and late-onset blind individuals show supra-normal auditory abilities in far-space. Curr Biol 2004 ; 14(19) : 1734-8.
[3] Rauschecker JP, Kniepert U. Auditory localization behavior in visually deprived cats. Eur J Neurosci 1994 ; 6(1) : 149-60.
[4] Röder B, Teder-Sälejärvi W, Sterr A, et al. Improved auditory spatial tuning in blind humans. Nature 1999 ; 400 : 162-6.
[5] Röder B, Stock O, Bien S, et al. Speech processing activates visual cortex in congenitally blind humans. Eur J Neurosci 2002 ; 16(5) : 930-6.
[6] Gougoux F, Lepore F, Lassonde M, et al. Neuropsychology : pitch discrimination in the early blind. Nature 2004 ; 430(6997) : 309.
[7] Niemeyer W, Starlinger I. Do the blind hear better ? Investigations on auditory processing in congenital or early acquired blindness. II. Central functions. Audiology 1981 ; 20(6) : 510-5.
[8] Bull R, Rathborn H, Clifford BR. The voice-recognition accuracy of blind listeners. Perception 1983 ; 12(2) : 223-6.
[9] Hugdahl K, Ek M, Takio F, et al. Blind individuals show enhanced perceptual and attentional sensitivity for identification of speech sounds. Brain Res Cogn Brain Res 2004 ; 19(1) : 28-32. Erratum in : Brain Res Cogn Brain Res 2004 ; 20(2) : 328.
[10] Raushecker JP, Korte M. Auditory compensation for early blindness in cat cerebral cortex. J Neurosci 1993 ; 13 : 4538-48.
[11] Korte T, Rauschecker JP. Auditory spatial tuning of cortical neurons is sharpened in cats with early blindness. J Neurophysiol 1993 ; 70 : 1717-21.
[12] Raushecker JP. Compensatory plasticity and sensory substitution in the cerebral cortex. TINS 1995 ; 18 : 36-43.
[13] Elbert T, Sterr A, Rockstroh B, et al. Expansion of the tonotopic area in the auditory cortex of the blind. J Neurosci 2002 ; 22(22) : 9941-4.
[14] Stevens AA, Snodgrass M, Schwartz D, Weaver K. Prepatory activity in occipital cortex in early blind humans predicts auditory perceptual performance. J Neurosci 2007 ; 27 : 10734-41.
[15] Wanet-Defalque MC, Veraart C, De Volder A, et al. High metabolic activity in the visual cortex of early blind human subjects. Brain Res 1998 ; 446 : 369-73.
[16] Gougoux F, Zatorre RJ, Lassonde M, et al. A functional neuro-imaging study of sound localization : visual cortex activity predicts performance in early-blind individuals. PLoS Biol 2005 ; 3(2) : e27.
[17] Collignon O, Renier L, Bruyer R, et al. Improved selective and divided spatial attention in early blind subjects. Brain Res 2006 ; 1075(1) : 175-82.
[18] Weeks R, Horwitz B, Aziz-Sultan A, et al. A positron emission tomographic study of auditory localization in the congenitally blind. J Neurosci 2000 ; 20(7) : 2664-72.
[19] Piché M, Robert S, Miceli D, Bronchti G. Environmental enrichment enhances auditory takeover of the occipital cortex in anophthalmic mice. Eur J Neurosci 2004 ; 20(12) : 3463-72.
[20] Pietrini P, Furey ML, Ricciardi E, et al. Beyond sensory images : object-based representation in the human ventral pathway. Proc Natl Acad Sci USA 2004 ; 101 : 5658-63.
[21] Collignon O, Lassonde M, Lepore F, et al. Functional cerebral reorganization for auditory spatial processing and auditory substituplustion of vision in early blind subjects. Cerebral Cortex 2007 ; 17(2) : 457-65.
[22] Renier LA, Anurova I, De Volder AG, et al. Multisensory integration of sounds and vibrotactile stimuli in processing streams for “What” and “Where”. J Neurosci 2009 ; 29 : 10950-60.
[23] Striem-Amit E, Ovadia-Caro S, Caramazza A, et al. Functional connectivity of visual cortex in the blind follows retinotopic organization principles. Brain 2015 ; 138(6) : 1679-95.
[24] Striem-Amit E, Dakwar O, Reich L, Amedi A. The large-scale organization of “visual” streams emerges without visual experience. Cerebral Cortex 2012 ; 22(7) : 1698-709.
[25] Hubel DH, Wiesel TN. The period of susceptibility to the physiological effects of unilateral eye closure in kittens. J Physiol 1970 ; 206(2) : 419-36.
[26] Lazzouni L, Lepore F. Compensatory plasticity : time matters. Front Neurosc 2014 ; 8 : A340.
[27] Chabot N, Charbonneau V, Laramée ME, et al. Sub-cortical auditory input to the primary visual cortex in anophthalmic mice. Neurosci Lett 2008 ; 433(2) : 129-34.
[28] Bach-y-Rita P, Collins CC, Saunders FA, et al. Vision substitution by tactile image projection. Nature 1969 ; 8 ; 221(5184) : 963-4.
[29] Capelle C, Trullemans C, Arno P, Veraart C. A real-time experimental prototype for enhancement of vision rehabilitation using auditory substitution. IEEE Trans Biomed Eng 1998 ; 45(10) : 1279-93.
[30] Maidenbaum S, Hanassy S, Abboud S, et al. The “EyeCane”, a new electronic travel aid for the blind : Technology, behavior & swift learning. Restor Neurol Neurosci 2014 ; 32(6) : 813-24.
[31] Buchs G, Maidenbaum S, Levy-Tzedek S, et al. Integration and binding in rehabilitative sensory substitution : Increasing resolution using a new zooming-in approach. Restor Neurol Neurosci 2016 ;34(1) : 97-105.
C. Meyniel
La « vision aveugle » ou « blindsight » en anglais est la persistance de fonctions visuelles inconscientes chez des personnes qui présentent une atteinte des aires visuelles. Elle est caractérisée par la capacité de localiser la position d’un stimulus présenté dans le champ visuel aveugle, voire de l’identifier, sans que ce stimulus ne soit consciemment perçu. Les patients avec une « vision aveugle » peuvent également suivre une cible, entraînant le mouvement des yeux en relation avec les mouvements de la scène visuelle.
De façon plus récente, des travaux ont mis en évidence la perception affective de la « vision aveugle ». Lors de la présentation de visages porteurs des différentes émotions, les patients sont capables de percevoir des émotions et ce de façon plus marquée lors de la peur ou du danger.
La « vision aveugle » peut être classée en deux catégories :
le type 1 est la capacité de discrimination visuelle en l’absence d’information consciente ;
alors que dans le type 2, le patient a une vague impression que quelque chose se passe.
Deux systèmes principaux sont impliqués dans la « vision aveugle ». Le premier, le système rétino-colliculo-thalamo-amygdalien, part de la rétine, passe par le colliculus supérieur et par le thalamus et se termine dans l’amygdale. Ce noyau est impliqué dans la perception des émotions. Le second, le système géniculo-extrastrié, part de la rétine, fait relais au niveau du corps géniculé latéral, pour se terminer directement dans les aires visuelles extrastriées, principalement dans l’aire V5. Cette dernière fait partie de la voie dorsale, ou voie du « où ? ». Aucun de ces deux systèmes ne passe par le cortex visuel primaire qui joue un rôle primordial dans la perception visuelle consciente. Des études en imagerie ainsi qu’en stimulation magnétique transcrânienne ont mis en évidence la réouverture de voies vestigiales chez ces patients telles qu’une communication directe entre les aires V5 droite et gauche [1, 2].
La réadaptation peut utiliser le blindsight dans les cécités non oculaires (hémianopsies, cécités corticales).
[1] Naccache L. Visual consciousness explained by its impairments. Curr Opin Neurol 2015 ; 28(1) : 45-50.
[2] Diederich NJ, Stebbins G, Schiltz C, Goetz CG. Are patients with Parkinson’s disease blind to blindsight ? Brain 2014 ; 137.
S. Picaud
Divers traumatismes ou pathologies de la rétine et du nerf optique se traduisent par la cécité des patients. Les patients concernés sont de l’ordre d’une vingtaine de millions dans le monde. Les prothèses rétiniennes ou corticales visent à redonner une perception visuelle utile à ces patients handicapés. Il s’agit de réintroduire une information visuelle dans le circuit neuronal. Cette intervention peut avoir lieu au niveau de la rétine si la pathologie résulte de la perte des photorécepteurs, les cellules qui transforment la lumière en une activité neuronale électrique. En effet, le réseau contient également deux couches de neurones dont les cellules bipolaires, postsynaptiques aux photorécepteurs, puis les cellules ganglionnaires qui transmettent l’information visuelle au cerveau. Ces deux couches neuronales transforment l’information visuelle sous forme analogique au niveau des photorécepteurs en un signal digital au niveau des cellules ganglionnaires.
Plus simplement, le potentiel membranaire du photorécepteur est une fonction linéaire de l’intensité lumineuse alors que le signal sera codé au niveau des cellules ganglionnaires sous la forme d’une fréquence de potentiel d’action. Ces deux couches neuronales persistant au moins partiellement, les prothèses rétiniennes pourront stimuler ces différentes couches neuronales pour envoyer un signal au cerveau.
Cette approche s’adresse aux patients atteints de pathologies des photorécepteurs comme les patients atteints de dégénérescence maculaire liée à l’âge (DMLA) ou de dystrophies héréditaires type rétinopathie pigmentaire (RP). Cependant, si la pathologie atteint directement les cellules ganglionnaires comme dans le glaucome ou la rétinopathie diabétique, les cellules ganglionnaires dégénèrent, rendant les prothèses rétiniennes inappropriées. Il en est de même pour les neuropathies optiques héréditaires ou alimentaires ainsi que pour les traumatismes du nerf optique.
Dans tous ces cas, les prothèses devraient être placées au niveau des aires visuelles supérieures pour induire une stimulation du cortex visuel ou du corps genouillé latéral dans le thalamus. Des essais cliniques prometteurs ont été réalisés avec les deux types d’implants soit rétiniens, soit corticaux. Les récents succès avec différentes prothèses rétiniennes démontrent le potentiel de ces approches de réhabilitation et devraient bénéficier également au développement et à la production de nouveaux implants thalamiques ou corticaux.
Après la perte des photorécepteurs, une prothèse rétinienne peut être placée : 1) dans l’espace sous-choroïdien ; 2) dans l’espace sous-rétinien à la place d’origine des photorécepteurs (implant sous-rétinien) ; 3) à la surface de la rétine à proximité des cellules ganglionnaires côté vitré (implant épirétinien) ; 4) sous forme de gaine autour du nerf optique. L’ensemble de ces implants peut produire une perception visuelle chez les patients aveugles, ce qui valide l’intérêt des approches de restauration visuelle. En effet, des études histopathologiques ont montré que des patients dans des stades avancés de rétinopathies pigmentaires pouvaient perdre jusqu’à deux tiers ou trois quart de leurs cellules ganglionnaires rétiniennes [1]. Cette perte neuronale s’accompagne d’un remodelage important de l’architecture cellulaire du tissu rétinien [2–4]. Il n’était donc pas certain que les cellules résiduelles dans un état de dégénérescence seraient encore en mesure de transmettre un signal au cerveau et si ce signal était toujours interprété comme une perception visuelle. Si les premiers tests cliniques en conditions aiguës ont permis de mettre en évidence la perception de phosphènes [5–7], les essais cliniques récents ont apporté la preuve d’une perception d’images interprétables par le cerveau [8–11]. Cependant, ces performances visuelles sont encore très variables suivant les patients et le niveau de vision atteint n’autorise pas encore la reconnaissance de visages ou les déplacements de manière autonome dans un environnement complexe.
Les développements précliniques en cours ont pour objectif d’atteindre de telles performances visuelles. Dans le cas des gaines d’électrodes autour du nerf optique, les phosphènes sont distribués dans tout le champ visuel sans pouvoir créer des images cohérentes, rendant cette approche difficilement exploitable par le patient [12]. Des approches transchoroïdiennes ont également été évaluées récemment en clinique [13–17]. Cependant, lorsque les patients localisent des objets ou des cibles lumineuses, leur acuité visuelle apparaît inférieure à ceux disposant d’implants rétiniens (voir ci-dessous).
Tous les dispositifs de ce type comportent les éléments suivants : 1) une caméra ou capteur optique situé dans une paire de lunettes pour acquérir les informations visuelles ; 2) un microprocesseur qui transforme les informations visuelles en codes de stimulations électriques ; 3) un émetteur qui transmet ces codes de stimulations vers le dispositif implanté ; 4) un dispositif attaché à l’oeil qui contient un ASIC (application-specific integrated circuit, puce électronique) transformant les codes de stimulations en véritables courants ; 5) sortant de ce dispositif, l’implant contenant les différentes pistes électriques conduisant aux électrodes à son extrémité qui est fixée à l’oeil et la rétine par un tack ou clou. Cet implant rentre dans l’oeil à la faveur d’une incision au niveau de l’ora serrata pour que les électrodes puissent être apposées à la surface de la rétine. La communication des codes de stimulations se fait par radiofréquence pour la société Second Sight (implant Argus 2®), alors qu’elle est assurée par faisceau infrarouge pour la société Pixium Vision (Implants IRIS®). La première société a finalisé ses essais cliniques internationaux avec un implant contenant 60 électrodes [10, 11], obtenant ainsi l’autorisation américaine de la Food and Drug Administration (FDA) et le marquage européen CE, puis bénéficiant en France du forfait innovation.
Dans un essai clinique multicentrique, 4 patients furent opérés au Centre hospitalier national d’ophtalmologie (CHNO) des Quinze-Vingts à Paris dans le service dirigé par le Pr Sahel. Avec ce dispositif, la majorité des patients pouvaient réaliser des tâches visuelles complexes telles que la localisation d’objets (96 % des sujets), la discrimination du mouvement (57 %) et la discrimination de réseaux orientés (23 %) [10]. La meilleure acuité visuelle mesurée était de 20/1260, encore six fois inférieure à la limite de la cécité légale (20/200). Deux des patients implantés ont également pu effectuer des tâches de lecture avec un taux pouvant aller jusqu’à 10 mots par minute.
La société Pixium Vision finalise actuellement ses essais cliniques internationaux multicentriques avec un dispositif à 49 électrodes. Les patients sont opérés à la Fondation ophtalmologique Rothschild dans le service du Pr Sahel, puis évalués fonctionnellement au CHNO des Quinze-Vingts dans le Centre d’investigation clinique en ophtalmologie. Les performances des patients semblent être sensiblement équivalentes avec le dispositif de l’entreprise Second Sight. En juin 2016, la société Pixium Vision a obtenu un marquage CE pour le dispositif IRIS 2®, équipé de 150 électrodes. Cette augmentation du nombre d’électrodes est importante, puisque des tests psychophysiques sur des personnes saines ont montré qu’un dispositif visuel avec 600 pixels pouvait permettre de lire un texte ou de se déplacer de manière autonome [18–20]. Cependant, il faut rappeler que chaque électrode stimulée ne produit pas automatiquement un pixel d’une image perçue.
Les deux dispositifs d’implants épirétiniens décrits ci-dessus se distinguent par le capteur optique, puisqu’il s’agit d’une simple caméra classique pour l’entreprise Second Sight, alors que la compagnie Pixium Vision dispose d’un capteur dit asynchrone. Ce capteur asynchrone ne fait pas d’images, mais chaque pixel mesure en temps réel les variations positives ou négatives d’intensité lumineuse dans sa direction de visée. L’absence de variation aboutit à l’absence totale de données, alors qu’une variation pourra être prise en compte à la milliseconde. Nous avons montré que les données issues de ce type de capteur peuvent permettre de modéliser toutes les réponses des cellules ganglionnaires avec une précision à la milliseconde [21]. Ce capteur est d’ailleurs souvent considéré comme un modèle in silico d’un type de cellules ganglionnaires de la rétine. Il faut également noter qu’un autre groupe a réalisé des essais cliniques confirmant la possibilité de restaurer une certaine perception visuelle avec un implant épirétinien [22].
Le dispositif Argus 2® stimule le tissu par des courants dont l’amplitude est une fonction linéaire de l’intensité du pixel considéré à une fréquence d’environ 20 Hz pour une durée d’impulsion de 100 μs à 1 ms, avec une stimulation biphasique, cathodique puis anodique. Cette stratégie fournit donc exactement les mêmes informations à tous les types de cellules ganglionnaires ne tenant pas compte de leur diversité [23]. Les cellules s’activant à l’allumage d’une lumière, cellules ON, recevront donc la même stimulation que celles qui s’activent à l’extinction de la phase lumineuse, cellules OFF. Outre la stimulation identique de toutes les cellules ganglionnaires de la rétine, l’approche épirétinienne induit également une stimulation des fibres ou axones des cellules ganglionnaires passant à la surface de la rétine en direction du nerf optique [7, 24]. Cette stimulation des fibres produit par conséquent une stimulation des cellules ganglionnaires plus périphériques et donc une perception d’un phosphène ayant une forme d’arc plus périphérique que le point dans le champ visuel correspondant à l’électrode. Il y a alors perte de la rétinotopie correspondant à la translation directe entre image projetée sur la rétine et perception de l’image. Ces différentes difficultés liées aux stimulations épirétiniennes pourraient expliquer la variabilité dans les performances des patients implantés avec l’Argus 2®, seulement 7 des 30 patients étant capables de réaliser de façon fiable les tâches d’acuité visuelle [10].
L’approche sous-rétinienne pourrait permettre de limiter ou supprimer ces différentes difficultés. L’entreprise Optobionics fut la première à évaluer de tels implants qui reposaient sur des photodiodes solaires directement insérées dans l’implant pour produire un courant de stimulation. Le dispositif devient donc très simple, puisque la puce électronique sous-rétinienne comprend les photodiodes et les électrodes. Aucun élément externe n’est nécessaire ; il faut simplement décoller la rétine résiduelle pour insérer dans l’espace sous-rétinien le dispositif circulaire de 2 mm de diamètre. Malheureusement, les photodiodes ne permettaient pas une conversion de suffisamment de photons pour entraîner l’activation du tissu rétinien. Seul un effet trophique à distance a pu être observé avec ce dispositif [25]. Pour résoudre ce problème technique, le Pr Zrenner et la société Retina Implant AG ont introduit un circuit amplificateur des signaux issus des photodiodes [26]. La puce reproduit donc exactement le fonctionnement d’un photorécepteur : réception des photons, amplification de ce signal, activation des neurones bipolaires. Cependant, la présence du circuit amplificateur impose l’addition d’une alimentation électrique avec un fil. Ce système rend par conséquent l’opération chirurgicale beaucoup plus complexe puisqu’elle oblige à insérer le dispositif dans l’espace sous-rétinien à partir de la périphérie jusqu’en position centrale. Dans un premier temps, le fonctionnement imparfait du dispositif a été compensé par la présence d’une matrice à 16 électrodes sous-rétiniennes reliées à l’extérieur par une liaison filaire, ce qui avait permis de démontrer l’obtention d’une perception visuelle sous l’effet d’une stimulation sous-rétinienne [8, 27].
Dans un second temps, le fonctionnement de la puce qui contient 1 500 électrodes a pu être testé de manière autonome, mais toujours avec l’alimentation électrique. Avec ce dispositif, les patients perçoivent des objets lumineux, discriminent des mires simples et peuvent dans certains cas lire les lettres. Retinal Implant rapporte une acuité visuelle de 20/1 000 chez ces patients [9]. Ces résultats peuvent paraître décevants puisque les performances visuelles sont du même ordre que celles des patients implantés avec l’Argus 2®, alors que les études psychophysiques estimaient que des implants à 600 pixels devraient permettre la reconnaissance des visages, la lecture de textes complexes et la locomotion autonome [18–20, 28, 29]. Cette incohérence avec la théorie pourrait s’expliquer par une diaphonie entre les électrodes voisines séparées de 70 μm qui partagent une électrode de masse très lointaine.
L’ensemble de ces travaux cliniques a apporté la preuve qu’un patient aveugle peut retrouver une vision utile avec une prothèse rétinienne. Cela signifie que le tissu peut encore transférer une information utile au cerveau malgré son engagement dans un processus neurodégénératif, même au niveau des cellules ganglionnaires. De plus, le cerveau peut interpréter ces informations visuelles comme des images. Ces résultats importants ont ouvert la voie vers la recherche de nouveaux procédés (prothèses ou thérapie optognétique). L’objectif pour les prothèses est d’augmenter la résolution tout en simplifiant le dispositif et donc la chirurgie associée.
Les travaux du Pr Zrenner et de l’entreprise Retina Implant AG ont démontré que le fait d’augmenter le nombre et la densité des électrodes ne repose pas sur la seule prouesse des électroniciens, mais qu’il faut sûrement repenser le design de la puce électronique pour que chaque électrode produise une stimulation rétinienne indépendante de sa voisine. Des travaux de modélisation ont montré que des stimulations bipolaires (une électrode de retour autour de l’électrode de stimulation) ou une configuration avec un plan de masse entourant toutes les électrodes pouvaient produire une bien meilleure sélectivité des stimulations que le mode actuel monopolaire (une électrode de retour distante commune pour toutes les électrodes de stimulations) [30]. C’est justement l’approche choisie par le groupe du Pr Palanker qui réintroduit le concept de photodiode, mais cette fois infrarouge avec un plan de masse local autour des électrodes de stimulation [31, 32]. Dans ce dispositif, chaque électrode est reliée à deux ou trois photodiodes en série, fournissant suffisamment de courant pour évoquer une activation du tissu rétinien. Les photodiodes sont sensibles à la lumière infrarouge qui devra donc être projetée par un projecteur monté dans une paire de lunettes et recevant les informations visuelles d’une caméra. Ce dispositif produit une acuité visuelle chez le rat inférieure d’un facteur 2 à l’acuité normale [33]. L’industrialisation de ces implants par la société Pixium Vision a permis leur évaluation à l’Institut de la Vision et la démonstration de leur efficacité sur des rétines ex vivo de primates non humains sans photorécepteurs.
L’utilisation de structures tridimensionnelles pourrait également améliorer la focalisation des stimulations [34–36]. De plus, l’introduction de matériaux innovants semi-conducteurs comme le diamant ou le graphène pourrait également augmenter la résolution des stimulations [37, 38], d’autant que nous avons montré leur biocompatibilité tant in vitro que in vivo [36, 39, 40].
Dans de nombreuses maladies (glaucome, rétinopathie diabétique, neuropathies optiques), la perte visuelle résulte de la perte des cellules ganglionnaires de la rétine qui communiquent l’information visuelle au cerveau. Dans ce cas, les prothèses rétiniennes ne peuvent pas restaurer la vision puisque le lien entre l’oeil et le cerveau est supprimé. La restauration visuelle implique par conséquent de réactiver les cellules des aires visuelles supérieures dans le thalamus ou le cortex visuel. Brindley et ses collaborateurs ont été les premiers à démontrer la possibilité de réactiver directement le cortex visuel [41]. Ensuite, des tests réalisés dans les années 1970-1980 ont montré une récupération fonctionnelle souvent transitoire avec des prothèses comprenant 100 électrodes [42, 43]. Le manque de persistance de la restauration visuelle a abouti à l’arrêt des essais cliniques dans l’attente d’une solution stable. Depuis cette période, les technologies ont complètement changé et de nouveaux dispositifs devraient prochainement rentrer en essais cliniques. En particulier, la société Second Sight a annoncé vouloir capitaliser sur son dispositif rétinien pour développer un dispositif cortical et procéder à des tests précliniques. Le succès des prothèses rétiniennes apporte de nouvelles briques pour le développement de ces prothèses corticales.
La restauration visuelle par des prothèses rétiniennes devient une approche réaliste, puisque certains patients ont montré des performances visuelles remarquables avec des implants contenant seulement 60 électrodes. Nous sommes sûrement au début de cette aventure qui nécessitera différentes améliorations des systèmes existants. Cependant, comme pour les implants cochléaires qui ont nécessité 20 ans de développement entre la reconnaissance d’un mot à la compréhension d’une phrase, nous pouvons espérer que des patients puissent d’ici quelques années retrouver une certaine autonomie dans leur déplacement, la reconnaissance visuelle de leurs proches ainsi que pour des tâches usuelles comme la lecture. L’innovation pour ces prothèses rétiniennes devrait faciliter le développement des prothèses corticales et la pérennité des stimulations produites par ce deuxième type de prothèse visuelle. Ces nouveaux résultats sont source d’espoir pour de nombreux patients aveugles suite aux pathologies les plus courantes1.
1. Ce texte a été relu par Laetitia Duhamel. Le projet évoqué est soutenu par l’Inserm, l’Université Pierre et Marie Curie (Paris VI), la Fondation ophtalmologique Adolphe de Rothschild (Paris), l’Agence nationale pour la recherche (ANR RETINE, ANR MEDINAS), le programme national français « Investissement d’Avenir » (LIFESENSES : ANR-10-LABX-65), la Banque publique d’investissement (projet SightAgain), la Fédération des aveugles de France, la Foundation Fighting Blindness, l’IRRP, la ville de Paris, le Conseil régional d’Île-de-France, la Commission européenne (projets DREAMS, NEUROCARE, flagship Graphène) et la Fondation pour la recherche médicale.
[1] Humayun MS, Prince M, de Juan E, Jr., et al. Morphometric analysis of the extramacular retina from postmortem eyes with retinitis pigmentosa. Invest Ophthalmol Vis Sci 1999 ; 40 : 143-8.
[2] Wang S, Villegas-Perez MP, Vidal-Sanz M, Lund RD. Progressive optic axon dystrophy and vacuslar changes in rd mice. Invest Ophthalmol Vis Sci 2000 ; 41 : 537-45.
[3] Marc RE, Jones BW, Watt CB, Strettoi E. Neural remodeling in retinal degeneration. Progress in Retinal and Eye Research 2003 ; 22 : 607-55.
[4] Marc RE, Jones BW, Anderson JR, et al. Neural reprogramming in retinal degeneration. Invest Ophthalmol Vis Sci 2007 ; 48 : 3364-71.
[5] Humayun MS, de Juan E, Jr., Dagnelie G, et al. Visual perception elicited by electrical stimulation of retina in blind humans. Arch Ophthalmol 1996 ; 114 : 40-6.
[6] Rizzo JF, 3rd, Wyatt J, Loewenstein J, et al. Methods and perceptual thresholds for short-term electrical stimulation of human retina with microelectrode arrays. Invest Ophthalmol Vis Sci 2003 ; 44 : 5355-61.
[7] Rizzo JF, 3rd, Wyatt J, Loewenstein J, et al. Perceptual efficacy of electrical stimulation of human retina with a microelectrode array during short-term surgical trials. Invest Ophthalmol Vis Sci 2003 ; 44 : 5362-9.
[8] Wilke R, Gabel VP, Sachs H, et al. Spatial resolution and perception of patterns mediated by a subretinal 16-electrode array in patients blinded by hereditary retinal dystrophies. Invest Ophthalmol Vis Sci 2011 ; 52 : 5995-6003.
[9] Zrenner E, Bartz-Schmidt KU, Benav H, et al. Subretinal electronic chips allow blind patients to read letters and combine them to words. Proceedings 2011 ; 278 : 1489-97.
[10] Humayun MS, Dorn JD, da Cruz L, et al. Interim results from the international trial of Second Sight’s visual prosthesis. Ophthalmology 2012 ; 119 : 779-88.
[11] da Cruz L, Coley BF, Dorn J, et al. The Argus II epiretinal prosthesis system allows letter and word reading and long-term function in patients with profound vision loss. Br J Ophthalmol 2013 ; 97 : 632-6.
[12] Veraart C, Raftopoulos C, Mortimer JT, et al. Visual sensations produced by optic nerve stimulation using an implanted self-sizing spiral cuff electrode. Brain Research 1998 ; 813 : 181-6.
[13] Fujikado T, Kamei M, Sakaguchi H, et al. Testing of semichronically implanted retinal prosthesis by suprachoroidal-transretinal stimulation in patients with retinitis pigmentosa. Invest Ophthalmol Vis Sci 2011 ; 52 : 4726-33.
[14] Morimoto T, Kamei M, Nishida K, et al. Chronic implantation of newly developed suprachoroidal-transretinal stimulation prosthesis in dogs. Invest Ophthalmol Vis Sci 2011 ; 52 : 6785-92.
[15] Villalobos J, Nayagam DA, Allen PJ, et al. A wide-field suprachoroidal retinal prosthesis is stable and well tolerated following chronic implantation. Invest Ophthalmol Vis Sci 2013 ; 54 : 3751-62.
[16] Ayton LN, Blamey PJ, Guymer RH, et al. Bionic Vision Australia C. First-in- human trial of a novel suprachoroidal retinal prosthesis. PloS One 2014 ; 9 : e115239.
[17] Nayagam DA, Williams RA, Allen PJ, et al. Chronic electrical stimulation with a suprachoroidal retinal prosthesis : a preclinical safety and efficacy study. PloS One 2015 ; 9 : e97182.
[18] Cha K, Horch KW, Normann RA. Mobility performance with a pixelized vision system. Vision Research 1992 ; 32 : 1367-72.
[19] Cha K, Horch KW, Normann RA, Boman DK. Reading speed with a pixelized vision system. J Opt Soc Am A 1992 ; 9 : 673-7.
[20] Sommerhalder J, Rappaz B, de Haller R, et al. Simulation of artificial vision : II. Eccentric reading of full-page text and the learning of this task. Vision Research 2004 ; 44 : 1693-706.
[21] Lorach H, Benosman R, Marre O, et al. Artificial retina : The multichannel processing of the mammalian retina achieved with a neuromorphic asynchronous light acquisition device. J Neural Eng 2012 ; 9 : 066004.
[22] Klauke S, Goertz M, Rein S, et al. Stimulation with a wireless intraocular epiretinal implant elicits visual percepts in blind humans. Invest Ophthalmol Vis Sci 2011 ; 52 : 449-55.
[23] Roska B, Werblin F. Vertical interactions across ten parallel, stacked representations in the mammalian retina. Nature 2001 ; 410 : 583-7.
[24] Wilms M, Eckhorn R. Spatiotemporal receptive field properties of epiretinally recorded spikes and local electroretinograms in cats. BMC Neuroscience 2005 ; 6 : 50.
[25] Chow AY, Chow VY, Packo KH, et al. The artificial silicon retina microchip for the treatment of vision loss from retinitis pigmentosa. Arch Ophthalmol 2004 ; 122 : 460-9.
[26] Zrenner E. Will retinal implants restore vision ? Science NY 2002 ; 295 : 1022-5.
[27] Besch D, Sachs H, Szurman P, et al. Extraocular surgery for implantation of an active subretinal visual prosthesis with external connections : feasibility and outcome in seven patients. Br J Ophthalmol 2008 ; 92 : 1361-8.
[28] Sommerhalder J, Oueghlani E, Bagnoud M, et al. Simulation of artificial vision : I. Eccentric reading of isolated words, and perceptual learning. Vision Research 2003 ; 43 : 269-83.
[29] Perez Fornos A, Sommerhalder J, Pittard A, et al. Simulation of artificial vision : IV. Visual information required to achieve simple pointing and manipulation tasks. Vision Research 2008 ; 48 : 1705-18.
[30] Joucla S, Yvert B. Improved focalization of electrical microstimulation using microelectrode arrays : a modeling study. PloS One 2009 ; 4 : e4828.
[31] Mathieson K, Loudin J, Goetz G, et al. Photovoltaic retinal prosthesis with high pixel density. Nat Photonics 2012 ; 6 : 391-7.
[32] Wang L, Mathieson K, Kamins TI, et al. Photovoltaic retinal prosthesis : implant fabrication and performance. J Neural Eng 2012 ; 9 : 046014.
[33] Lorach H, Goetz G, Smith R, et al. Photovoltaic restoration of sight with high visual acuity. Nat Med 2015 ; 21 : 476-82.
[34] Palanker D, Huie P, Vankov A, et al. Migration of retinal cells through a perforated membrane : implications for a high-resolution prosthesis. Invest Ophthalmol Vis Sci 2004 ; 45 : 3266-70.
[35] Djilas M, Oles C, Lorach H, et al. Three-dimensional electrode arrays for retinal prostheses : modeling, geometry optimization and experimental validation. J Neural Eng 2011 ; 8 : 046020.
[36] Bendali A, Rousseau L, Lissorgues G, et al. Synthetic 3D diamond-based electrodes for flexible retinal neuroprostheses : Model, production and in vivo biocompatibility. Biomaterials 2015 ; 67 : 73-83.
[37] Hadjinicolaou AE, Leung RT, Garrett DJ, et al. Electrical stimulation of retinal ganglion cells with diamond and the development of an all diamond retinal prosthesis. Biomaterials 2012 ; 33 : 5812-20.
[38] Kiran R, Rousseau L, Lissorgues G, et al. Multichannel boron doped nanocrystalline diamond ultramicroelectrode arrays : Design, fabrication and characterization. Sensors 2012 ; 12 : 7669-81.
[39] Bendali A, Agnes C, Meffert S, et al. Distinctive glial and neuronal interfacing on nanocrystalline diamond. PloS One 2014 ; 9 : e92562.
[40] Bendali A, Hess LH, Seifert M, et al. Purified neurons can survive on peptide-free graphene layers. Adv Healthc Mater 2013 ; 2 : 929-33.
[41] Brindley GS, Lewin WS. The sensations produced by electrical stimulation of the visual cortex. J Physiol 1968 ; 196 : 479-93.
[42] Dobelle WH, Mladejovsky MG, Girvin JP. Artifical vision for the blind : electrical stimulation of visual cortex offers hope for a functional prosthesis. Science NY 1974 ; 183 : 440-4.
[43] Dobelle WH. Artificial vision for the blind by connecting a television camera to the visual cortex. Asaio J 2000 ; 46 : 3-9.
C.P. Hamel
Au fur et à mesure de la découverte des gènes responsables de dystrophie rétinienne, il est apparu que les protéines codées par ces gènes peuvent être regroupées en fonction du métabolisme dans lequel elles sont impliquées, offrant ainsi un éclairage d’ensemble sur les causes de ces maladies. Cette classification présente un intérêt physiopathologique et permet aussi d’identifier des caractéristiques cliniques communes.
Divers aspects de la physiologie des photorécepteurs sont impliqués, au premier rang desquels on trouve les processus qui sont spécifiques à la vision comme la transduction visuelle, en étroite relation avec les processus de régénération des pigments visuels (cycle visuel). Des gènes intervenant dans l’épissage des ARNm (acide ribonucléique messager), la synthèse protéique et le trafic cellulaire, le cytosquelette des segments externes et le développement des photorécepteurs sont aussi en cause, ainsi que ceux codants certaines protéines de matrices extracellulaires ou de l’épithélium pigmentaire de la rétine (EPR).
La transduction visuelle est une chaîne de réactions déclenchées par la lumière, qui s’effectue dans le segment externe du photorécepteur et qui conduit à la production du message nerveux visuel. Beaucoup des effecteurs de cette chaîne sont impliqués dans les dystrophies rétiniennes, en particulier dans la rétinite pigmentaire (RP) (Fig. 9-2). La mise en jeu de la chaîne de transduction commence par l’isomérisation par la lumière du chromophore de la rhodopsine, le rétinal 11-cis, en rétinal tout-trans, ce qui active la rhodopsine et permet la liaison de la sous-unité alpha de la transducine Après l’échange de la guanosine diphosphate (GDP) en guanosine triphosphate (GTP), la transducine alpha se fixe sur la sous-unité gamma de la phosphodiestérase, désinhibant ainsi les sous-unités catalytiques alpha et bêta, ce qui fait chuter le taux de guanosine monophosphate cyclique (GMPc) et entraîne la fermeture des canaux ioniques, conduisant à l’hyperpolarisation du photorécepteur et à la diminution de libération de glutamate. La réaction est arrêtée par la re-synthèse du GMPc grâce à une guanylate cyclase, mais aussi par la désensibilisation de la rhodopsine effectuée par la phosphorylation du site de liaison à la transducine grâce à une rhodopsine kinase, puis par la liaison de l’arrestine sur le site phosphorylé, et enfin par l’hydrolyse de la GTP liée à la transducine alpha.
Les mutations de la rhodopsine sont en majorité responsables de RP à hérédité autosomique dominante alors que, pour la plupart des autres protéines, l’hérédité est autosomique récessive. Il s’agit des gènes codant les sous-unités alpha et bêta de la phosphodiestérase et les sous-unités alpha et bêta du canal ionique couplé à la GMPc. Dans le cas de la phosphodiestérase, la perte de fonction va entraîner d’une part des taux de GMPc importants, toxiques pour la cellule, et d’autre part le maintien des canaux à l’état constamment ouvert, obligeant la cellule à un fonctionnement intensif de la pompe Na/K+–ATPase qui la mène à un épuisement métabolique. De plus, l’excès de calcium intracellulaire résultant de l’ouverture constante des canaux conduit à l’altération de la perméabilité des membranes mitochondriales, qui déclenche l’activation de l’apoptose. Dans le cas des mutations du canal ionique couplé à la GMPc, la perte de fonction entraîne au contraire une fermeture permanente du canal, voire son absence d’adressage à la membrane plasmique. L’absence de canaux ioniques fonctionnels, équivalente à l’activation constitutive des photorécepteurs, conduirait à la dégénérescence de ces derniers par l’activation constante des mécanismes de désactivation induits par le faible taux de calcium. Ainsi, la faible concentration de calcium activerait la guanylate cyclase, conduisant, comme dans le cas de mutations de la phosphodiestérase (PDE), à des taux de GMPc toxiques pour le photorécepteur. On voit donc que, bienque ces différents effecteurs fassent partie d’une même chaîne de signalisation, le type de transmission de la maladie et les mécanismes physiopathologiques en cause sont très différents.
Notons que des mutations d’autres gènes de la transduction visuelle sont impliquées dans d’autres dystrophies rétiniennes, comme la guanylate cyclase, responsable de l’amaurose congénitale de Leber et de cone-rod dystrophies, et comme l’activateur de guanylate cyclase 1, responsable de dystrophies des cônes, de cone-rod dystrophies et de maculopathies. Dans le même ordre d’idée, des mutations de la transducine alpha sont responsables d’héméralopie essentielle de type Nougaret, de même que certaines mutations de la rhodopsine et de la phosphodiestérase peuvent aussi donner des héméralopies essentielles.
À la fin du processus de transduction visuelle, le rétinal tout-trans se libère de l’opsine. Afin de rendre l’opsine réactivable sous forme de rhodopsine, il faut un apport nouveau de rétinal 11-cis. La synthèse de cet isomère de la vitamine A s’effectue dans l’EPR par un processus pluri-enzymatique appelé cycle visuel (Fig. 9-3). Dans le cytoplasme du segment externe, le rétinal tout-trans est réduit en rétinol tout-trans par une déshydrogénase, traverse la matrice interphotoréceptrice grâce à l’interstitial retinol binding protein (IRBP) et rejoint l’EPR où il est estérifié par la lecithin retinol acyl transferase (LRAT). C’est alors qu’intervient RPE65, une enzyme qui va assurer l’hydrolyse de l’ester et l’isomérisation en rétinol 11-cis. Ce dernier est pris en relais par la cellular retinaldehyde binding protein (CRALBP) et est oxydé par une déshydrogénase spécifique des formes cis (RDH5). Le rétinal 11-cis obtenu est repris en charge par l’IRBP et aboutit au photorécepteur, où il peut se combiner avec l’opsine pour reformer de la rhodopsine. Enfin, une dernière protéine localisée dans la membrane des disques des segments externes, ABCA4, joue un rôle dans le métabolisme du rétinol, en transportant, de la lumière du disque vers le cytoplasme, un complexe associant rétinal tout-trans et phosphatidyl éthanolamine (PE), le N-rétinylidène- PE, mais aussi sans doute du rétinal tout-trans. Le transport de ce PE constitue une véritable détoxification, évitant ainsi la formation du N-rétinylidène- N- rétinyléthanolamine (A2-E), un composant essentiel de la lipofuscine.
De nombreux gènes codant des protéines du cycle visuel sont en cause dans les dystrophies rétiniennes. Des mutations de la CRALBP conduisent à la rétinite ponctuée albescente. Des mutations de la LRAT et surtout de RPE65, donnent des formes particulières d’amaurose congénitale de Leber marquées par un besoin de lumière très important. Toutes ces mutations agissent par perte de fonction et les dystrophies rétiniennes qui en découlent se transmettent sur le mode autosomique récessif. Le mécanisme pathogénique, surtout étudié pour RPE65, mais probablement similaire pour LRAT et CRALBP, passe par le faible niveau de rhodopsine, lié au manque de rétinal 11-cis. Lorsque ce niveau est très bas, le photorécepteur contient beaucoup d’opsine libre capable d’activer en permanence la transducine, conduisant ainsi à la dégénérescence lente des photorécepteurs. Des mutations d’ABCA4 sont responsables de maladie de Stargardt, mais si les deux allèles du gène conduisent à l’absence totale de fonction, on peut observer une véritable rétinite pigmentaire. Ici, le mécanisme physiopathologique est très différent, car il passe par une accumulation de lipofuscine dans l’EPR et une dégénérescence des photorécepteurs secondaire à une pathologie de l’EPR. D’autres gènes impliqués dans le métabolisme du rétinol sont responsables de formes diverses de dystrophies rétiniennes. Ainsi, des mutations de RDH5 causent le fundus albipunctatus.
Bien d’autres gènes exprimés dans les photorécepteurs peuvent donner des dystrophies rétiniennes. Mentionnons le groupe de facteurs contrôlant l’épissage des ARNm qui ont pour particularité de donner des RP dominantes non syndromiques avec des taux de pénétrance très variables, certains sujets porteurs pouvant ne présenter aucun signe de la maladie ; le groupe des protéines intervenant dans la formation du cil connecteur et la vectorisation protéique dans le cil, qui sont la cause de diverses formes de RP, mais aussi d’amauroses congénitales de Leber et de diverses formes syndromiques de dystrophies rétiniennes (syndrome de Bardet-Biedl, syndrome de Joubert), ainsi que du syndrome d’Usher, la forme de RP syndromique la plus fréquente.

Fig. 9-2 Activation de la transduction visuelle des bâtonnets.
Les principaux effecteurs de la transduction visuelle sont représentés. Ceux qui sont en rouge sont impliqués dans des dystrophies rétiniennes (voir le texte). CNG : canal ionique couplé à la guanosine monophosphate cyclique (GMPc), sous-unités A1 ou B1 ; GCAP : guanylate cyclase activating protein ; PDE : phosphodiestérase ; RetGC : retinal guanylate cyclase ; RHO : rhodopsine.
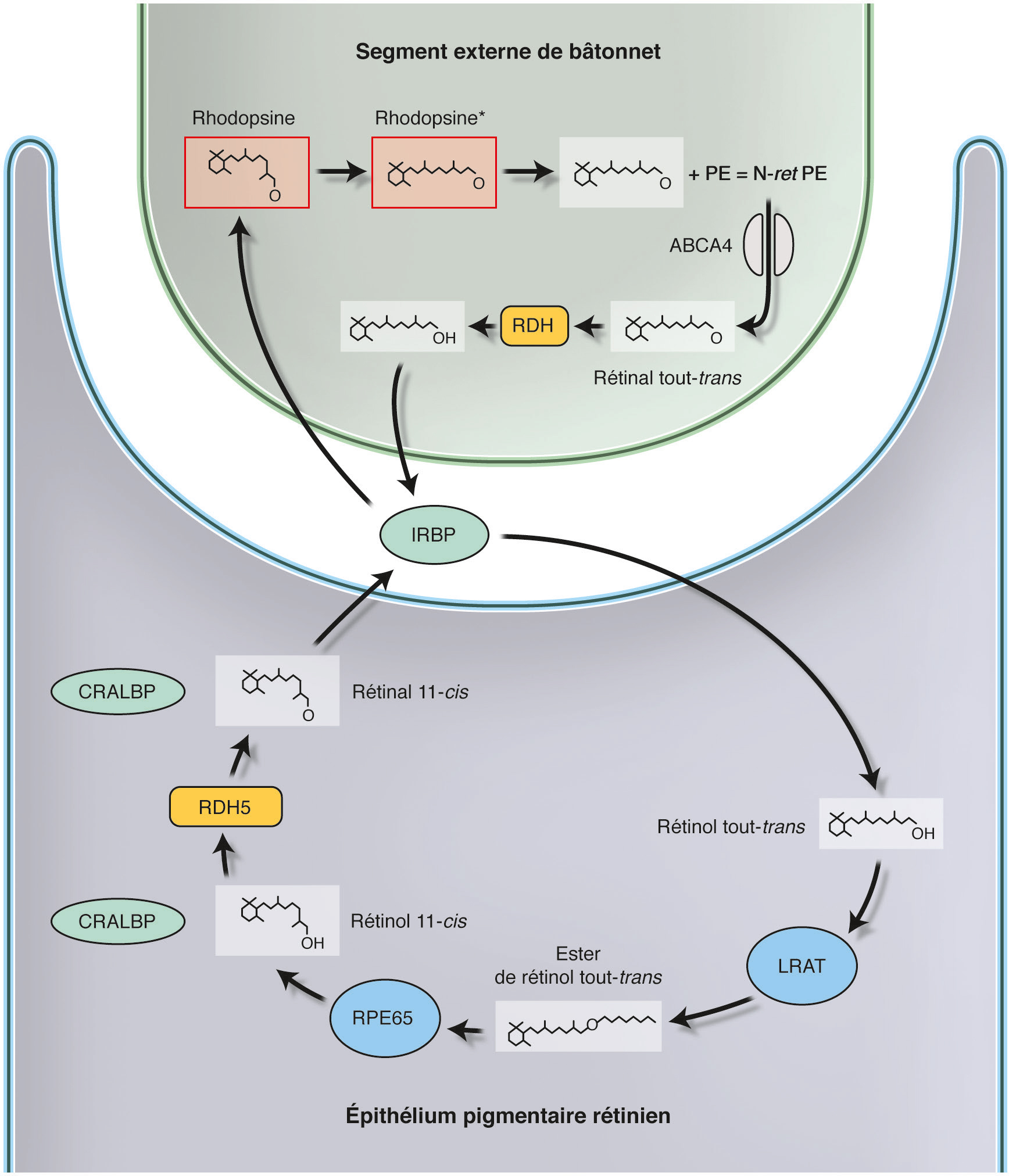
Fig. 9-3 Cycle visuel.
Voir le texte pour les commentaires. ABCA4 : ATP-binding cassette A4 ; CRALBP : cellular retinaldehyde binding protein ; IRBP : interphotoreceptor retinol binding protein ; LRAT : lecithin retinol acyl transferase ; OH : hydroxyle ; PE : phosphatidyl éthanolamine ; RDH : retinol dehydrogenase.
G. Le Meur, M. Weber
La thérapie génique, qui est l’introduction dans une cellule cible d’un gène thérapeutique, est un espoir pour le traitement des dégénérescences rétiniennes héréditaires, comme les rétinopathies pigmentaires, et celles non héréditaires, comme la dégénérescence maculaire liée à l’âge (DMLA). Ces dernières années, nous avons vu, enfin, arriver l’application de cette thérapeutique, qui semblait jusqu’alors confiné aux laboratoires, à la clinique au cours des premiers essais chez l’homme. Depuis plusieurs années, plus d’une dizaine d’essais cliniques de phase I à III ont été menés. Les premiers essais cliniques ont concerné les dystrophies rétiniennes héréditaires liées à une mutation du gène RPE65 [1–4] avec une probable autorisation de mise sur le marché (AMM) dans les années à venir. Depuis, il y a eu d’autres essais cliniques dans d’autres pathologies : la choroïdérémie liée X, les dystrophies rétiniennes liées au gène MERTK, la maladie de Stargardt, l’achromatopsie, le syndrome d’Usher de type 1B, le rétinoschisis lié X et l’atrophie optique de Leber (eTableau 9-1). Nous présenterons, ici, les résultats publiés concernant les essais liés aux mutations du gène RPE65 et à la choroïdérémie liée X.
Les mutations du gène RPE65 sont responsables d’amaurose congénitale de Leber et de rétinopathies pigmentaires survenant précocement dans l’enfance. Six essais, de phases I/II à III, concernant cette pathologie ont été menés avec un recul de plus de 6 ans pour certains pour un total de 94 patients (eTableau 9-1). Un des essais a d’ailleurs testé une réadministration dans l’oeil controlatéral chez 3 patients [5]. Les vecteurs utilisés ont été un vecteur de type AAV de sérotype 2 ou de sérotype 4 avec soit le promoteur RPE65 spécifique des cellules de l’épithélium pigmenté [1] soit un promoteur ubiquitaire [2–5]. Les volumes d’injection sous-rétinienne ont varié de 150 μl à 1 ml [2–5]. Ces essais cliniques ont démontré, en ce qui concerne le traitement par thérapie génique, une absence d’effets délétères généraux.
Une réponse immune minime et non délétère a été rapportée par Maguire [6] et Jacobson [7]. Une faible réponse immune sans effet clinique a été rapportée lors de la réadministration dans l’oeil controlatéral [5]. Au niveau ophtalmologique, il a été rapporté l’apparition d’un trou lamellaire suite à l’injection sous-rétinienne chez un patient [2]. Ce problème serait lié à la présence préopératoire d’une membrane épirétinienne (MER) chez ce patient. Par la suite, les patients qui présentaient une MER ont d’abord été opérés de celle-ci avant la réalisation de l’injection sous-rétinienne. Une équipe a décrit la survenue chez 3 patients d’une inflammation clinique intraoculaire moyenne et transitoire pour les patients ayant reçu la dose du vecteur la plus forte [8]. En ce qui concerne les modifications des capacités visuelles, il a été noté une amélioration de la fixation avec une diminution du nystagmus après le traitement [9], des gains d’acuité visuelle variables [6, 10], avec, pour certains, une stabilité à 3 ans [9]. Une amélioration du champ visuel a été notée pour certains, notamment pour les patients les plus jeunes [6, 8]. Une amélioration de la cinétique pupillaire a été publiée par deux équipes (Fig. 9-4a) ainsi qu’une amélioration de la sensibilité rétinienne (Fig. 9-4b) [6, 10]. Ces modifications sont aussi liées à une amélioration des déplacements des patients, avec une diminution du temps de déplacement et une diminution du nombre de percussions lors d’un parcours d’obstacles chronométré (Fig. 9-4c) [8, 11]. Une équipe a publié l’amélioration de l’IRM fonctionnelle lors d’une stimulation haut contraste à la suite du traitement [12]. En revanche, dernièrement, deux équipes ont rapporté une diminution des bénéfices fonctionnels passé 3 ans de suivi [8, 13]. D’ailleurs, Jacobson rapporte une diminution de l’épaisseur rétinienne mesurée par SD-OCT (spectral domain optical coherence tomography) à 3 ans post-injection, avec néanmoins déjà pour certains patients une diminution dès la première année [13]. Les prochaines années vont être importantes afin de savoir quelles seront les modalités d’applications de ce traitement, que ce soit en termes de durée d’efficacité de traitement ou en termes de dose, de geste chirurgical, car les modifications d’épaisseur rétinienne pourraient être liées au décollement fovéolaire.

Fig. 9-4 Modifications fonctionnelles après traitement par thérapie génique RPE65 chez des patients RPE65-/-.
a. Modification de la cinétique pupillaire pour un patient selon Maguire [6]. b. Modification de la sensibilité rétinienne mesurée au champ visuel (FST) avec stimulation bleue entre la mesure préopératoire et le contrôle à 3 ans selon Jacobson [10]. c. Modification des capacités de déplacements 6 mois après traitement en fonction de la luminosité selon Bainbridge [8].
En 2014, l’équipe de MacLaren a publié les résultats concernant l’essai de phase I-II réalisé dans la choroïdérémie liée à X [14]. Six patients, âgés de 35 à 63 ans, ont été opérés par injection sous-rétinienne de 0,1 ml du vecteur AAV.REP1 à la dose de 1 × 1010 vecteur génome (vg), sauf pour le patient 6 (0,6 × 1010 vg). Aucun effet secondaire général ou ophtalmologique n’a été dénoté au cours de cet essai clinique. L’épaisseur rétinienne, mesurée à l’OCT, avant et 6 mois après injection, est restée stable pour les 6 patients suite à l’injection sous-rétinienne. Deux patients ont vu leur acuité visuelle augmenter de 21 et 11 lettres sur l’échelle ETDRS (Early Treatment Diabetic Retinopathy Study) (Fig. 9-5a). La sensibilité rétinienne a augmenté en moyenne de 2,3 dB lors du contrôle à 6 mois de la chirurgie pour les yeux traités, contrairement aux yeux non traités (–0,8 dB). L’augmentation de sensibilité semble être liée à la dose de vecteur injectée (Fig. 9-5b). Ce premier essai, qui a inclus des patients fortement atteints, semble être prometteur.
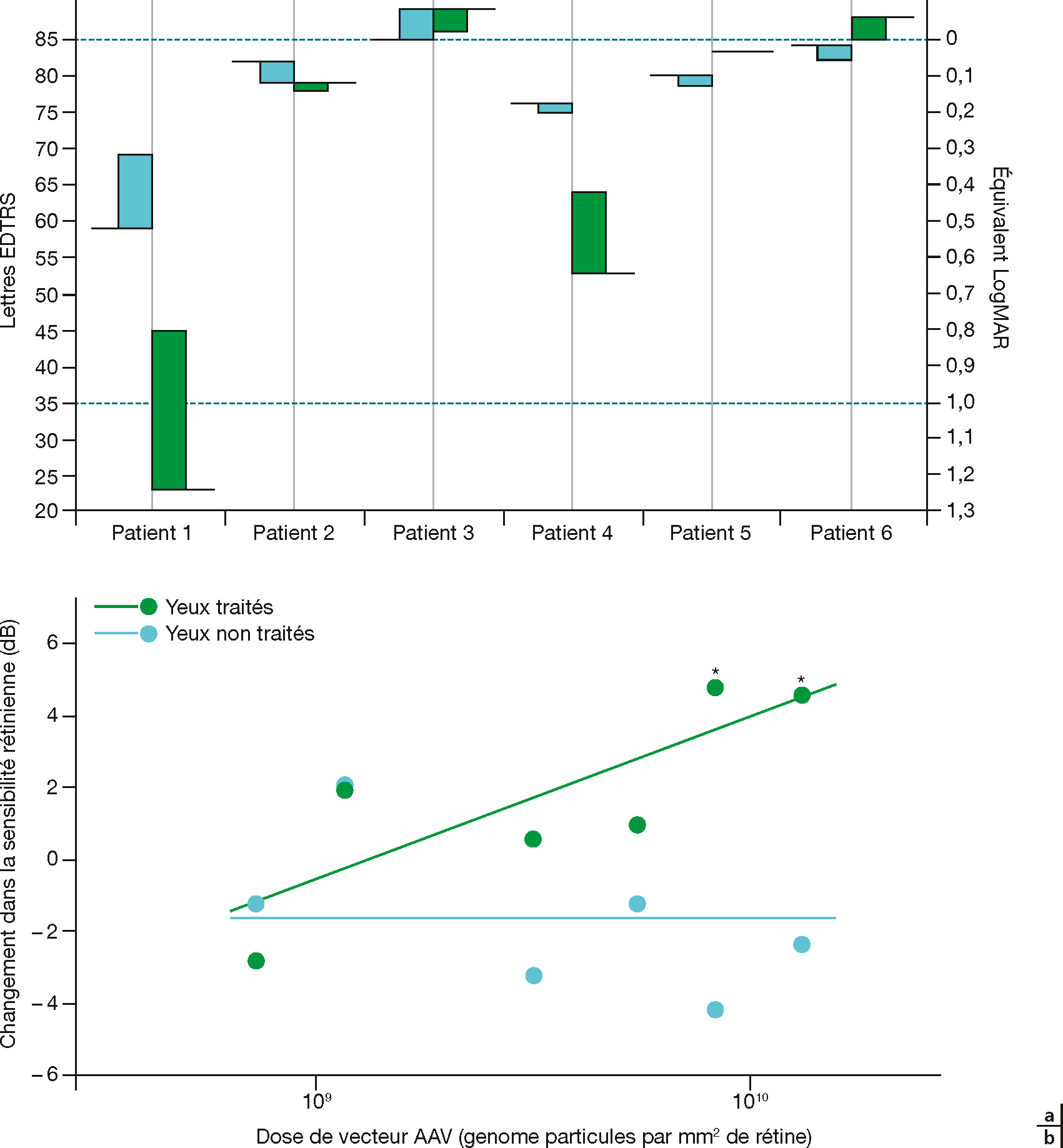
Fig. 9-5 Modification visuelle et doses de vecteurs administrées chez les patients atteints de choroïdérémie selon MacLaren [14].
a. Modification dans l’acuité visuelle à échelle ETDRS pour chacun des 6 patients de l’étude de MacLaren. Les traits horizontaux représentent le niveau basal et les colonnes les modifications 6 mois après le traitement pour les yeux traités (vert) et les yeux non traités (bleu). b. Modification de la sensibilité rétinienne mesurée par micropérimétrie avant et 6 mois après le traitement dans les yeux traités et dans les yeux contrôles. L’amélioration dans les yeux traités est corrélée à la dose de vecteur génome par mm2 de rétine viable.
La thérapie génique des maladies héréditaires ophtalmologiques est un réel espoir pour de nombreux patients. Les divers essais cliniques concernant le gène RPE65 ou le gène REP1 ont démontré la sécurité de tels traitements dans ces pathologies rétiniennes. L’amélioration de la fonction rétinienne sera à évaluer dans le temps afin de s’assurer de la stabilité de ce traitement par transfert de gène. Dans les prochaines années, la communauté ophtalmologique sera le témoin de ces avancées thérapeutiques que nous pourrons probablement proposer à nos patients.
[1] Bainbridge JW, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med 2008 ; 358(21) : 2231-9.
[2] Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med 2008 ; 358(21) : 2240-8.
[3] Cideciyan AV, Hauswirth WW, Aleman TS, et al. Human RPE65 gene therapy for Leber congenital amaurosis : persistence of early visual improvements and safety at 1 year. Hum Gene Ther 2009 ; 20(9) : 999-1004.
[4] Banin E, Bandah-Rozenfeld D, Obolensky A, et al. Molecular anthropology meets genetic medicine to treat blindness in the North African jewish population : human gene therapy initiated in Israel. Hum Gene Ther 2010 ; 6.
[5] Bennett J, Ashtari M, Wellman J, et al. AAV2 gene therapy readministration in three adults with congenital blindness. Sci Transl Med 2012 ; 4(120) : 120ra15.
[6] Maguire AM, High KA, Auricchio A, et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis : a phase 1 dose-escalation trial. Lancet 2009 ; 374(9701) : 1597-605.
[7] Cideciyan AV, Hauswirth WW, Aleman TS, et al. Human RPE65 gene therapy for Leber congenital amaurosis : persistence of early visual improvements and safety at 1 year. Hum Gene Ther 2009 ; 20(9) : 999-1004.
[8] Bainbridge JW, Mehat MS, Sundaram V, et al. Long-term effect of gene therapy on Leber’s congenital amaurosis. N Engl J Med 2015 ; 372(20) : 1887-97.
[9] Testa F, Maguire AM, Rossi S, et al. Three-year follow-up after unilateral subretinal delivery of adeno-associated virus in patients with Leber congenital amaurosis type 2. Ophthalmology 2013 ; 120(6) : 1283-91.
[10] Jacobson SG, Cideciyan AV, Ratnakaram R, et al. Gene therapy for leber congenital amaurosis caused by RPE65 mutations : safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol 2012 ; 130(1) : 9-24.
[11] Simonelli F, Maguire AM, Testa F, et al. Gene therapy for Leber’s congenital amaurosis is safe and effective through 1.5 years after vector administration. Mol Ther 2010 ; 18(3) : 643-50.
[12] Ashtari M, Cyckowski LL, Monroe JF, et al. The human visual cortex responds to gene therapy-mediated recovery of retinal function. J Clin Invest 2011 ; 121(6) : 2160-8.
[13] Jacobson SG, Cideciyan AV, Roman AJ, et al. Improvement and decline in vision with gene therapy in childhood blindness. N Engl J Med 2015 ; 372(20) : 1920-6.
[14] MacLaren RE, Groppe M, Barnard AR, et al. Retinal gene therapy in patients with choroideremia : initial findings from a phase 1/2 clinical trial. Lancet 2014 ; 383 : 1129-37.
Auricchio A, Rolling F. Adeno-associated viral vectors for retinal gene transfer and treatment of retinal diseases. Curr Gene Ther 2005 ; 5(3) : 339-48.
Chévez-Barrios P, Chintagumpala M, Mieler W, et al. Response of retinoblastoma with vitreous tumor seeding to adenovirus-mediated delivery of thymidine kinase followed by ganciclovir. J Clin Oncol 2005 ; 23(31) : 7927-35.
Rasmussen H, Chu KW, Campochiaro P, et al. Clinical protocol. An open-label, phase I, single administration, dose-escalation study of ADGVPEDF.11D (ADPEDF) in neovascular age-related macular degeneration (AMD). Hum Gene Ther 2001 ; 12(16) : 2029-32.
S. Picaud
Le succès des essais cliniques sur les prothèses rétiniennes (voir chap. 9-3) a montré que réactiver la rétine de certains patients aveugles peut induire une perception visuelle. La thérapie optogénétique propose de redonner une vision utile à de tels patients devenus aveugles par le transfert d’un gène microbien (algue, bactérie). Ce gène microbien code pour une protéine photosensible, soit une opsine-canal ionique, soit une opsine-transporteur ionique. L’activation de la protéine par la lumière produit un courant à travers la membrane dans laquelle la protéine est intégrée. En conséquence, son expression dans la membrane cellulaire d’un neurone transforme cette cellule en un véritable photorécepteur. La production du courant ionique dans la membrane permet de contrôler optiquement le potentiel de membrane du neurone. Par conséquent, après la perte des photorécepteurs ou leur inactivation, les neurones résiduels de la rétine devraient pouvoir être resensibilisés à la lumière pour restaurer une perception visuelle. Cette forme de thérapie génique non conventionnelle devrait pouvoir s’appliquer aussi bien aux patients atteints de maladies rares d’origines génétiques comme la rétinopathie pigmentaire, mais aussi à ceux atteints de maladies plus complexes comme la dégénérescence maculaire liée à l’âge (DMLA). Il s’agit d’une thérapie génique, puisque le gène est introduit dans les neurones résiduels de la rétine par des vecteurs viraux de type adéno-associés (adeno-associated virus [AAV]). Cependant, le gène n’est pas naturellement présent dans le patrimoine génétique humain, mais provient d’un micro-organisme, algue ou bactérie. Ces mécanismes microbiens de photoréception peuvent être considérés comme relativement archaïques ; la contrepartie de leur simplicité est la nécessité de très fortes luminances pour les activer, la protéine ne disposant pas des mécanismes d’amplification présents dans les photorécepteurs humains. Cette thérapie optogénétique est actuellement en cours d’évaluation sur les primates non humains et devrait prochainement entrer en clinique.
Ce chapitre décrit donc les différentes formes de thérapie optogénétique qui sont actuellement en cours d’étude au niveau préclinique. Il aborde également des stratégies de restauration visuelle fondées sur d’autres opsines voire sur des agents pharmacologiques photosensibilisants.
Si la thérapie génique classique est en plein essor suite aux succès du traitement de l’amaurose congénitale de Leber, elle ne peut pas encore cibler tous les patients atteints de dystrophies rétiniennes soit que l’origine génétique ne soit pas connue, soit qu’il soit déjà trop tard. Pour les patients atteints de rétinopathie pigmentaire, la cause génétique est identifiée dans la moitié des cas. Cependant, la thérapie génique ne peut pas s’appliquer dans les formes dominantes, puisque la mutation introduit un gain de fonction résultant en une toxicité. Enfin, la thérapie génique doit évidemment être pratiquée sur des photorécepteurs en bon état, donc bien avant leur entrée dans un processus de dégénérescence. Pour les patients atteints de DMLA, si des prédispositions génétiques ont été identifiées comme facteur de risque de la maladie, la thérapie génique n’est pas encore considérée pour ces patients. Or, actuellement, seules les complications vasculaires peuvent être traitées chez les patients atteints de DMLA, mais aucun traitement ne cible la dégénérescence des photorécepteurs, hormis les compléments alimentaires.
De nombreux patients atteints de DMLA ou de dystrophies rétiniennes sont donc déjà aveugles ou évoluent vers la cécité. Comme pour les prothèses rétiniennes, les premiers patients qui devraient être inclus dans les essais cliniques de la thérapie optogénétique sont atteints de dystrophies rétiniennes (par exemple rétinopathie pigmentaire). La conservation d’un champ visuel périphérique chez les patients atteints de DMLA les exclut de ces essais tant que le bénéfice ne permet pas d’atteindre des performances visuelles nettement supérieures.
Dans les photorécepteurs, une fois activées par la lumière, les opsines peuvent activer la transducine, une protéine G qui active elle-même une phosphodiestérase qui clive à son tour la guanosine monophosphate cyclique (GMPc), réduisant ainsi sa concentration. La GMPc contrôlant l’ouverture d’un canal ionique, son hydrolyse va aboutir à la réduction du courant dit d’obscurité qui dépolarise les photorécepteurs. Une opsine activée peut activer plusieurs transducines et une phosphodiestérase peut également cliver une grande partie de la GMPc. Ces deux mécanismes d’amplification sont finement régulés par différentes protéines et par le calcium intracellulaire. Ils permettent au bâtonnet d’atteindre une gamme de sensibilité pouvant aller jusqu’à la détection d’un seul photon. Si les opsines-canal et opsines-transporteur contiennent, comme les opsines humaines, un chromophore dérivé de la vitamine A, le fonctionnement est différent, puisque le couplage avec le canal ou le transporteur est direct au sein de la même protéine.
La bactériorhodopsine fut la première protéine de ce type étudiée dans les années 1970. Elle transporte les protons alors que l’halorhodopsine décrite un peu plus tard transporte des ions chlorures [1]. C’est la découverte de la protéine dénommée channelrhdopsin2 dans l’algue Chlamydomonas reinhardtii qui a véritablement lancé la thérapie optogénétique, car ses découvreurs montraient que son expression permettait de contrôler le potentiel membranaire de cellules de mammifères [2]. Son application au déclenchement d’une activité neuronale sous forme de potentiels d’actions voire de comportement fut apportée dans la foulée [3, 4]. Si cette protéine déclenche l’activité, d’autres opsines peuvent rendre un neurone silencieux sous l’effet de la lumière [5, 6]. Depuis, différents variants ont permis d’augmenter la sensibilité à la lumière ou le temps d’ouverture de cette protéine [7, 8]. Il s’agit en particulier de la protéine CatCh dont la sensibilité à la lumière a été augmentée par un facteur 70 en augmentant sa perméabilité au calcium. Par ailleurs, la channelrhodopsin-2 étant sensible à la lumière bleue, certains ont développé des variants ou isolé de nouvelles opsines-canal comme ChrimsonR, avec une sensibilité spectrale dans le rouge, moins toxique pour les cellules et avec une meilleure pénétration dans les tissus [9–11].
Dans la rétine, le traitement de l’information se divise selon deux canaux ON et OFF qui s’activent et s’inhibent respectivement en présence de lumière. Les différents protagonistes ont par conséquent proposé d’activer la voie ON par la protéine channerlhodopsine2 et d’inhiber la voie OFF par la protéine halorhodopsine. Le premier à utiliser la thérapie optogénétique pour réactiver la rétine de souris aveugles fut le Pr Zhao Pan de Détroit [12]. Il montrait sur la souris rd1 que l’expression de la channelrhodopsin dans les cellules ganglionnaires permet de réactiver ces cellules lors d’une stimulation lumineuse. Une approche similaire fut également appliquée sur le rat RCS, permettant d’enregistrer des réponses corticales voire des réponses comportementales [13, 14]. Pour évaluer un potentiel transfert clinique, le Pr Pan a examiné le taux de transfection dans un primate non humain, le marmouset [15]. Ce travail montre que des cellules ganglionnaires peuvent effectivement être activées directement par la channelrhodopsin2. Cependant, l’oeil de marmoset est bien plus petit que celui de l’homme et les vecteurs viraux AAV ne présentent pas le même pattern de transfection que dans des espèces comme les macaques, plus proches phylogénétiquement de l’homme.
Enfin, l’expression d’une protéine d’algue dans l’oeil humain pose un questionnement majeur sur la réaction immunitaire que pourraient déclencher les cellules recouvertes de cette protéine. C’est pourquoi, à l’Institut de la Vision, nous (Deniz Dalkara, Jens Duebel, José Sahel, Serge Picaud) avons évalué l’effet de la thérapie optogénétique sur la rétine du macaque. Pour cette expérience, nous avons choisi d’utiliser la protéine Catch, de plus grande sensibilité à la lumière, et de la placer sous un promoteur spécifique des cellules ganglionnaires pour limiter l’expression dans d’autres types cellulaires. Les résultats montrent une expression sélective de la protéine dans les cellules ganglionnaires d’un anneau périfovéolaire. Cette restriction de l’expression avait déjà été décrite par D. Dalkara, même avec les vecteurs AAV sélectionnés dans un processus d’évolution dirigée [16]. L’enregistrement en patch-clamp permet de démontrer la production de courants photo-induits dans les cellules ganglionnaires de l’anneau périfovéolaire, alors que leur enregistrement sur matrice d’électrodes démontre la production de potentiels d’action en réponse avec une stimulation lumineuse dont l’intensité serait compatible avec une stimulation chez l’homme. Aucune réaction inflammatoire majeure n’est à dénoter. Par conséquent, ces résultats ouvrent la voie pour une étude de toxicité de plus grande ampleur avant le lancement d’essais cliniques.
Si cette approche ciblant les cellules ganglionnaires pourrait prochainement être évaluée en clinique, d’autres voies alternatives sont également considérées. Le ciblage des cellules ganglionnaires par la channelrhodopsine2 ou ses variants aboutit à une réponse ON dans toutes les cellules ganglionnaires, même a priori dans les cellules OFF. Inversement, l’expression de l’halorhodopsine peut supprimer l’activité spontanée des cellules indifféremment de leur statut individuel ON ou OFF. Le précédent des prothèses rétiniennes suggère que les patients devraient pouvoir interpréter correctement ces signaux, mais seuls les patients pourront expliquer la perception produite par la stimulation directe et indifférenciée de ces cellules ganglionnaires. Cependant, B. Roska a proposé de cibler les cellules bipolaires de type ON en utilisant un promoteur spécifique [17]. Les souris rd1 aveugles retrouvent une perception visuelle et des réponses ON sont produites dans les cellules ganglionnaires. Ces expériences ayant été faites par électroporation, l’expérience a été reproduite par d’autres avec des vecteurs viraux [18]. Plus récemment, nous avons repris cette étude avec d’autres vecteurs viraux spécifiquement manipulés pour faciliter leur pénétration dans la rétine [16]. Nous avons alors montré que l’activation des cellules bipolaires ON permet de produire des réponses ON et OFF tant sur la rétine qu’au niveau du cortex visuel [19]. Cette activation des circuits ON et OFF tient au transfert physiologique de l’activité des cellules bipolaires ON à bâtonnet dans les circuits ON et OFF des cônes. Malheureusement, nous ne disposons pas actuellement de vecteurs viraux capables de transduire efficacement les cellules bipolaires chez les primates.
La découverte au cours de ces expériences d’un nombre important de photorécepteurs résiduels a amené B. Roska à suggérer qu’il serait possible de réactiver ces photorécepteurs dormants. En effet, ces photorécepteurs ne possèdent plus de segment interne/ externe leur permettant de répondre à une stimulation lumineuse. Pour reproduire l’hyperpolarisation induite par la lumière, l’expression de la protéine halorhodopsine, qui produit une hyperpolarisation des neurones lors de la stimulation optique, a été obtenue dans les photorécepteurs de la souris rd1. Le résultat est surprenant : des fonctions visuelles aussi complexes que la détection des mouvements sont à nouveau mesurables au niveau des cellules ganglionnaires de la rétine [20]. Développant la rétine humaine postmortem en culture [21], nous avons pu montrer avec l’équipe retide Roska que l’halorhodopsine s’exprime dans les photorécepteurs humains et qu’elle y est fonctionnelle [20]. L’intérêt thérapeutique de cette approche reposait sur la présence de tels photorécepteurs dits « dormants » chez des patients aveugles. L’examen par tomographie de cohérence optique (OCT) de leur rétine a permis de confirmer la présence de tels photorécepteurs dormants chez certains patients aveugles suite à une rétinopathie pigmentaire [20]. La réactivation de leur rétine devrait donc être possible par l’activation de ces photorécepteurs dormants. Cependant, des premiers essais d’expression de l’halorhodopsine chez le primate non humain vivant n’ont pas permis d’atteindre un niveau d’expression suffisant pour prendre le contrôle optique de ses photorécepteurs. Par conséquent, dans l’état actuel de nos connaissances, le ciblage des cellules ganglionnaires semble l’approche optogénétique la plus prometteuse.
Le succès des prothèses rétiniennes et de la thérapie optogénétique a stimulé des recherches alternatives très originales. Il a par exemple été proposé d’exprimer la rhodopsine dans les cellules bipolaires de type ON [22]. Cette expression ectopique permet de retrouver une réponse à la lumière dans les cellules ganglionnaires de la souris rd1. Ce phénomène reposerait sur la capacité des récepteurs à protéines G d’interagir dans leurs différents mécanismes de transduction du signal. Ainsi, la rhodopsine qui interagit directement avec la transducine, une protéine G, pourrait également interagir avec la protéine Go qui intervient dans la cascade de transduction du récepteur métabotropique au glutamate, mGluR6, des cellules bipolaires ON [23]. La sensibilité de ce mécanisme à la GMPc avait déjà montré certaines analogies [24], même s’il comporte d’importantes différences [24]. Cependant, la cinétique des réponses est lente et incompatible en l’état avec une utilisation clinique. L’avantage de ces approches serait d’utiliser des séquences de protéines naturellement présentes chez l’homme et donc moins susceptibles de déclencher des réactions immunitaires. Cette réflexion avait également justifié l’utilisation de la mélanopsine, le pigment des cellules ganglionnaires intrinsèquement sensibles à la lumière [25], qui avait été exprimé dans tout type de cellules ganglionnaires de la rétine [26]. Cependant, les constantes cinétiques de cette protéine apparaissent incompatibles avec l’induction de réponse dans une dynamique suffisamment rapide pour la perception visuelle [26]. Pour augmenter l’efficacité de ces activations par des opsines naturelles, une chimère de mélanopsine et de récepteur mGluR6 a été produite et exprimée dans les cellules bipolaires de type ON. Cette expression par thérapie génique permet d’induire des réponses ON et OFF à la lumière dans les cellules ganglionnaires de la souris rd1 [27].
Enfin, une approche purement pharmacologique a également été proposée pour resensibiliser la rétine de patients aveugles. L’idée repose sur l’injection de molécules non toxiques qui pourraient déclencher l’ouverture de canaux ioniques sous l’effet d’une stimulation lumineuse. Cette approche a actuellement été validée sur la rétine ex vivo et/ou in vivo de souris ou rats aveugles [28, 29]. Elle présente l’inconvénient de nécessiter des injections de molécules photosensibilisantes au niveau de l’oeil à intervalles de temps réguliers. Ces injections régulières sont un inconvénient sur le long terme, mais elles pourraient présenter l’avantage de pouvoir interrompre rapidement le processus en cas de problème majeur.
L’approche optogénétique ouvre de nouvelles perspectives pour la restauration visuelle de patients aveugles ou malvoyants. Le ciblage des cellules ganglionnaires rétiniennes devrait être la première approche en clinique d’ici 1 à 2 ans, une fois l’absence de toxicité et de réponse immunitaire évaluée. Cependant, de nouvelles stratégies alternatives pourraient également entrer en clinique dans les dix prochaines années. Un succès de la thérapie optogénétique en ophtalmologie pourrait ouvrir le champ des possibles pour cette approche dans des maladies neurodégénératives et le traitement du handicap. En effet, la validation de la thérapie génique en ophtalmologie et la présence d’une fenêtre optique sur la rétine font de la restauration visuelle l’application la plus aisée et la plus pertinente pour la thérapie optogénétique. Les essais cliniques devraient prochainement apporter la réponse à la question de son efficacité chez les patients.
[1] Bamberg E, Tittor J, Oesterhelt D. Light-driven proton or chloride pumping by halorhodopsin. Proc Natl Acad Sci U S A 1993 ; 90(2) : 639-43.
[2] Nagel G, Szellas T, Huhn W, et al. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci U S A 2003 ; 100(24) : 13940-5.
[3] Boyden ES, Zhang F, Bamberg E, et al. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci 2005 ; 8(9) : 1263-8.
[4] Nagel G, Brauner M, Liewald JF, et al. Light activation of channelrhodopsin- 2 in excitable cells of Caenorhabditis elegans triggers rapid behavioral responses. Curr Biol 2005 ; 15(24) : 2279-84.
[5] Chow BY, Han X, Dobry AS, et al. High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature 2010 ; 463(7277) : 98-102.
[6] Cosentino C, Alberio L, Gazzarrini S, et al. Optogenetics. Engineering of a light-gated potassium channel. Science 2015 ; 348(6235) : 707-10.
[7] Kleinlogel S, Feldbauer K, Dempski RE, et al. Ultra light-sensitive and fast neuronal activation with the Ca(2)+-permeable channelrhodopsin CatCh. Nat Neurosci 2011 ; 14(4) : 513-8.
[8] Pan ZH, Ganjawala TH, Lu Q, et al. ChR2 mutants at L132 and T159 with improved operational light sensitivity for vision restoration. PLoS One 2014 ; 9(6) : e98924.
[9] Zhang F, Prigge M, Beyriere F, et al. Red-shifted optogenetic excitation : a tool for fast neural control derived from Volvox carteri. Nat Neurosci 2008 ; 11(6) : 631-3.
[10] Lin JY, Knutsen PM, Muller A, et al. ReaChR : a red-shifted variant of channelrhodopsin enables deep transcranial optogenetic excitation. Nat Neurosci 2013 ; 16(10) : 1499-508.
[11] Klapoetke NC, Murata Y, Kim SS, et al. Independent optical excitation of distinct neural populations. Nat Methods 2014 ; 11(3) : 338-46.
[12] Bi A, Cui J, Ma YP, et al. Ectopic expression of a microbial-type rhodopsin restores visual responses in mice with photoreceptor degeneration. Neuron 2006 ; 50(1) : 23-33.
[13] Tomita H, Sugano E, Fukazawa Y, et al. Visual properties of transgenic rats harboring the channelrhodopsin-2 gene regulated by the thy-1.2 promoter. PLoS One 2009 ; 4(11) : e7679.
[14] Tomita H, Sugano E, Isago H, et al. Channelrhodopsin-2 gene transduced into retinal ganglion cells restores functional vision in genetically blind rats. Exp Eye Res 2010 ; 90(3) : 429-36.
[15] Ivanova E, Hwang GS, Pan ZH, Troilo D. Evaluation of AAV-mediated expression of Chop2-GFP in the marmoset retina. Invest Ophthalmol Vis Sci 2010 ; 51(10) : 5288-96.
[16] Dalkara D, Byrne LC, Klimczak RR, et al. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retidenal gene delivery from the vitreous. Sci Transl Med 2013 ; 5(189) : 189ra176.
[17] Lagali PS, Balya D, Awatramani GB, et al. Light-activated channels targeted to ON bipolar cells restore visual function in retinal degeneration. Nat Neurosci 2008 ; 11(6) : 667-75.
[18] Doroudchi MM, Greenberg KP, Liu J, et al. Virally delivered channelrhodopsin-2 safely and effectively restores visual function in multiple mouse models of blindness. Mol Ther 2011 ; 19(7) : 1220-9.
[19] Mace E, Caplette R, Marre O, et al. Targeting channelrhodopsin- 2 to on-bipolar cells with vitreally administered AAV restores on and off visual responses in blind mice. Mol Ther 2015 ; 23(1) : 7-16.
[20] Busskamp V, Duebel J, Balya D, et al. Genetic reactivation of cone photoreceptors restores visual responses in retinitis pigmentosa. Science 2010 ; 329(5990) : 413-7.
[21] Fradot M, Busskamp V, Forster V, et al. Gene therapy in ophthalmology : validation on cultured retinal cells and explants from postmortem human eyes. Hum Gene Ther 2011 ; 22(5) : 587-93.
[22] Cehajic-Kapetanovic J, Eleftheriou C, Allen AE, et al. Restoration of Vision with Ectopic Expression of Human Rod Opsin. Curr Biol 2015 ; 25(16) : 2111-22.
[23] Nawy S. The metabotropic receptor mGluR6 may signal through G(o), but not phosphodiesterase, in retinal bipolar cells. J Neurosci 1999 ; 19(8) : 2938-44.
[24] de la Villa P, Kurahashi T, Kaneko A. L-glutamate- induced responses and cGMP-activated channels in three subtypes of retinal bipolar cells dissociated from the cat. J Neurosci 1995 ; 15(5 Pt 1) : 3571-82.
[25] Hattar S, Liao HW, Takao M, et al. Melanopsin-containing retinal ganglion cells : architecture, projections, and intrinsic photosensitivity. Science 2002 ; 295(5557) : 1065-70.
[26] Lin B, Koizumi A, Tanaka N, et al. Restoration of visual function in retinal degeneration mice by ectopic expression of melanopsin. Proc Natl Acad Sci U S A 2008 ; 105(41) : 16009-14.
[27] van Wyk M, Pielecka-Fortuna J, Lowel S, Kleinlogel S. Restoring the on switch in blind retinas : opto-mGluR6, a next-generation, cell-tailored optogenetic tool. PLoS Biol 2015 ; 13(5) : e1002143.
[28] Polosukhina A, Litt J, Tochitsky I, et al. Photochemical restoration of visual responses in blind mice. Neuron 2012 ; 75(2) : 271-82.
[29] Tochitsky I, Polosukhina A, Degtyar VE, et al. Restoring visual function to blind mice with a photoswitch that exploits electrophysiological remodeling of retinal ganglion cells. Neuron 2014 ; 81(4) : 800-13.