

H. Dollfus, K. Chennen
Seuls 5 % des glaucomes primitifs à angle ouvert (GPAO) sont monogéniques (résultant d’une mutation dans un seul gène) et généralement de transmission autosomique dominante.
Deux gènes sont principalement impliqués dans les formes monogéniques : la myociline et l’optineurine.
Pour 95 % des GPAO, les facteurs génétiques sont des facteurs de prédisposition qui s’additionnent entre eux et à des facteurs environnementaux.
Les études d’association pour le GPAO sont nombreuses et parfois contradictoires.
L’étude d’endophénotypes (sous-catégories cliniques) est très utile pour affiner l’étude des facteurs de risque.
Les facteurs de prédisposition génétique du GPAO sont mieux identifiés, leur connaissance est en plein essor, avec des implications cliniques surprenantes et le développement du rôle des facteurs épigénétiques.
Le GPAO représente une des grandes causes de cécité à travers le monde et a ainsi motivé de nombreuses équipes à en déterminer les facteurs génétiques. La composante génétique du GPAO est connue de longue date. En effet, une histoire familiale de GPAO est souvent présente dans ces familles [13], et les études familiales ont montré une augmentation de 10 % du risque de développer un GPAO pour des apparentés du premier degré d’un individu atteint [31]. Une autre étude a montré que des apparentés d’une personne atteinte ont, au cours de leur vie, 22 % de risque de développer un GPAO, alors que le risque pour des apparentés de personnes qui ne sont pas atteintes de glaucome est de 2 à 3 % [31]. Malgré cette forte composante génétique, il a été identifié de manière convaincante des facteurs clairement liés à cette maladie, seulement dans 5 % de l’ensemble de la population des personnes atteintes de GPAO. Ainsi, pour les 95 % restants de cette population, il existe encore des facteurs génétiques qui restent à déterminer clairement malgré de nombreux travaux parfois contradictoires, car l’identification de ces facteurs génétiques s’avère plus compliquée que prévu.
D’une manière générale, le GPAO peut être considéré comme une maladie dite « commune » et « complexe » sur le plan génétique tout en tenant compte de facteurs environnementaux qui jouent aussi un rôle majeur. En effet, il n’existe que très peu de familles qui correspondent à une transmission mendélienne classique, à savoir qu’une mutation dans un gène donné est suffisante et nécessaire pour que la maladie apparaisse (fig. 4-1).
A contrario, dans la majorité des cas de GPAO, des facteurs génétiques, qui sont certainement nombreux, sont plus difficiles et plus subtils à identifier et nécessitent des approches génétiques de type GWAS (genome wide association studies – études d’associations pangénomiques) sur de grandes cohortes de patients. Ce type d’étude a été utilisé pour identifier les facteurs génétiques dans d’autres pathologies communes, telles que l’asthme par exemple, l’hypertension artérielle ou la dégénérescence maculaire liée à l’âge. Pour cette dernière, des facteurs génétiques ont été identifiés avec succès, notamment ceux qui ont été rapportés aux variants du facteur H du complément [8, 14, 17], éclairant d’un jour nouveau la physiopathogénie si complexe de cette affection. Des études identiques ont été effectuées pour la dystrophie endothéliale de Fuchs avec des variants dans le gène TCF4 [2] et les syndromes exfoliatifs avec des variants identifiés par exemple dans le gène LOXL1 [29]. L’intérêt d’identifier ces facteurs génétiques ou de prédisposition génétique réside dans une meilleure compréhension physiopathogénique de l’affection en mettant en évidence de nouvelles protéines impliquées dans ces maladies, et permettant ainsi d’identifier des facteurs de risque chez les patients. À ce jour, il faut considérer le GPAO comme une maladie complexe sur le plan génétique, impliquant très probablement plusieurs, voire de nombreuses sous-catégories de patients. Dans l’évaluation de pathologies complexes, l’utilisation d’endophénotypes ou encore de traits quantitatifs est souvent utilisée pour identifier des facteurs qui pourraient aider dans l’analyse des cohortes. C’est le cas pour le GPAO, notamment en ce qui concerne par exemple le ratio C/D (CDR) vertical [3, 5], la pression intra-oculaire (PIO) [5], l’épaisseur centrale de la cornée (ECC) [5] ou la surface du disque optique [20]. Ces endophénotypes ont été particulièrement utilisés récemment dans les études d’association sur diverses populations à travers le monde, car ils permettent de déterminer des sous-catégories de cohorte plus ciblées pour les analyses type GWAS.
Ainsi, d’une manière générale dans les facteurs héréditaires du GPAO, deux catégories de gènes sont reconnues : les gènes mendéliens, qui sont peu nombreux et concernent une minorité de patients, et les gènes de facteurs de prédisposition associés qui sont, eux, très nombreux et concernent une majorité de patients, mais de manière beaucoup plus subtile.
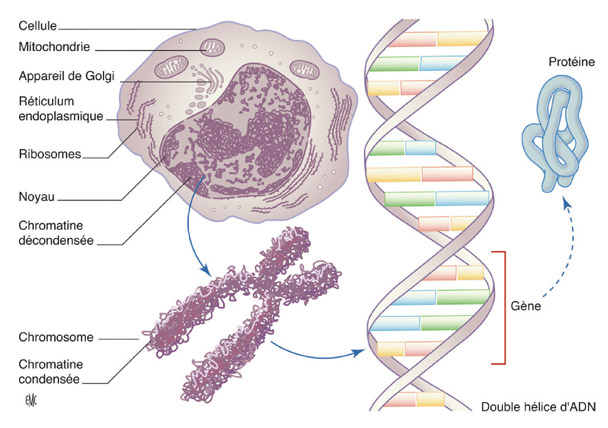
Fig. 4-1 Représentation schématique de la cellule et du noyau contenant les chromosomes, formés en partie d’ADN. Les gènes sont situés le long des chromosomes et codent pour une protéine. (Source : Dollfus H, Pelletier, V. EMC Ophthalmologie, 2008 ; 5 : 1-14.)
Les mutations dans les gènes mendéliens sont situées au niveau d’un seul gène par individu ; ils ont un effet important chez cet individu pour enclencher l’apparition de la maladie, et dépendent ici très peu de l’intervention d’autres gènes ou de l’environnement. Dans le GPAO, il s’agit le plus souvent d’hérédité à transmission autosomique dominante, c’est-à-dire qu’elle touche un seul allèle d’un individu, et un individu atteint (hétérozygote) porteur de la mutation va avoir 50 % de risque de transmission à sa descendance ; l’arbre généalogique sera dit vertical, c’est-à-dire avec des cas présents de génération en génération, touchant aussi bien les hommes que les femmes (fig. 4-2). Deux gènes sont des exemples classiques de cette situation : le gène de la myociline (MYOC) et celui de l’optineurine (OPTN).
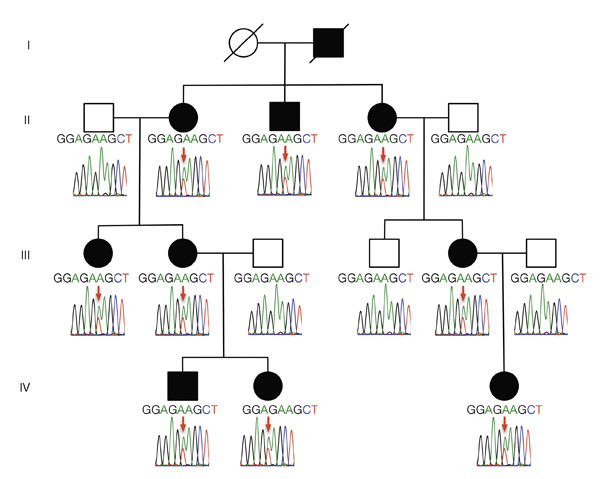
Fig. 4-2 Représentation de la transmission d’un glaucome autosomique dominant au sein d’une famille sur quatre générations (I-IV). Les individus de phénotype sain ont des formes blanches et ceux ayant un glaucome sont représentés par une forme pleine en noir. L’allèle muté (A → T) est représenté par la flèche en rouge sur un électrophérogramme résultant d’un séquençage automatique.
En 1997, l’équipe de Stone et Sheefield, aux États-Unis, identifie un gène responsable de GPAO dans une grande famille d’origine caucasienne autosomique dominante et habitant aux États-Unis. Vingt-deux membres de cette famille étaient atteints de glaucome juvénile à angle ouvert et tous étaient porteurs d’une mutation unique dans le gène de la myociline (MYOC) [28]. Le rôle exact de cette protéine au niveau du trabéculum, de l’humeur aqueuse et de l’élévation importante de la PIO qu’entraînent ces perturbations génétiques reste l’objet de nombreuses discussions [19]. Il n’en demeure pas moins que ce gène est responsable, d’une manière générale dans le monde, de 3 à 5 % des GPAO. Plus de 70 mutations ont été rapportées [16], la plus fréquente étant la mutation Q368X, qui a d’ailleurs été plutôt associée à des formes de début très précoce. Les mutations semblent particulièrement impliquées dans des formes avec des PIO très élevées, aussi bien dans les formes précoces que tardives de l’affection. Compte tenu de l’importance de la mutation G368X dans de nombreuses ethnies (en dehors de l’Asie), il semble qu’il pourrait y avoir un ancêtre commun. Des modèles animaux portant des mutations dans la myociline sont à l’étude pour identifier les facteurs moléculaires caractéristiques de l’élévation de la PIO dans le glaucome lié à la myociline.
Le deuxième gène responsable de forme monogénique de GPAO a été identifié grâce à l’étude d’une très grande famille atteinte de glaucome à pression normale, avec une transmission autosomique dominante et porteuse de la mutation p.Glu50Lys (ou E50K) dans l’optineurine [27]. À l’inverse des mutations dans le gène de la myociline, celles-ci semblent plutôt limitées à certaines populations et la contribution génétique est plus complexe à analyser [10].
Sur le plan physiopathogénique, l’optineurine a été étudiée dans des expériences in vitro et in vivo, montrant un rôle de neuroprotection pour ce gène ; des souris transgéniques porteuses de la mutation p.Glu50Lys ont démontré une apoptose des cellules ganglionnaires rétiniennes et une implication potentielle avec le transport cellulaire protéique au niveau de ces cellules [7].
D’autres gènes ont été identifiés par des approches identiques aux deux précédents, mais néanmoins sont sujets à controverse dans la littérature. Nous les mentionnons brièvement puisqu’ils font toujours l’objet de travaux de recherche.
Pour le gène TANK binding kinase 1 (TBK1), des variations du nombre de copie (des duplications) ont été identifiées par analyse de liaison dans une large famille afro-américaine [11]. De manière intéressante, ce gène interagit avec l’optineurine et est impliqué dans la voie NF-κB (nuclear factor-kappa B) et pourrait moduler l’apoptose via le système immunitaire [11, 23]. Des études complémentaires sont néanmoins nécessaires pour évaluer l’importance de ce gène dans le GPAO.
Le gène WD repeat domaine 36 (WDR36) a également été identifié à travers l’analyse de grandes familles de glaucome, et des mutations dans ce gène seraient à l’origine de GPAO. L’association GPAO avec WDR36 n’a pas été retrouvée aux États-Unis, en Australie, au Canada et en Allemagne [12]. Néanmoins, ce gène est l’objet de nombreuses controverses dans la littérature, étant considéré par certains plutôt comme un gène contribuant aux facteurs de risque de développer des formes polygéniques de glaucome ou d’en influencer sa sévérité [9, 15, 22].
Une étude européenne a montré que des mutations dans le gène NFT4 codant pour la neurotrophine 4 pouvaient être trouvées chez 1,7 % des patients [25]. Ces résultats sont également sujets à controverse, notamment en fonction de l’origine géographique, et ce gène ne concernerait finalement qu’une minorité de patients [30].
Les études génétiques d’association concernent les maladies complexes comme le GPAO. Elles sont en général utilisées pour de grandes cohortes avec une étude de type cas-témoins, et sont élaborées pour explorer l’intégralité du génome à la recherche de variants génétiques associés à la maladie. Les SNP (single nucleotide polymorphism – polymorphisme nucléotidique) sont étudiés à travers tout le génome dans les cas patients et chez les sujets témoins ; la fréquence des allèles (variation au niveau d’une base donnée) est alors comparée entre les deux groupes (fig. 4-3).
La stratégie est fondée sur l’hypothèse d’un déséquilibre de liaison, c’est-à-dire qu’il y a un assortiment d’allèles assez proches entre eux sur un chromosome (donc qui ne recombineront pas), qui sont différents entre le groupe des cas et le groupe de témoins. Cette approche peut identifier des zones génomiques associées à la maladie et nécessite ensuite des études de génotypage et fonctionnelles approfondies pour être validées et confirmées sur un plan biologique. Ensuite, ce type d’analyse doit être répliqué sur des cohortes indépendantes et faire l’objet d’études élargies avec des méta-analyses.
De manière générale, ces approches ont été très utilisées dans ces dix dernières années. Elles ont aussi été très commentées dans la littérature quant à leur interprétabilité car elles ne peuvent pas vraiment détecter d’héritabilité, phénomène dénommé par les Anglo-Saxons « the missing heritability » [21]. Il n’en demeure pas moins que des variations génétiques (dans des séquences codantes ou non) existent dans des régions bien déterminées du génome dénommées locus (fig. 4-4).
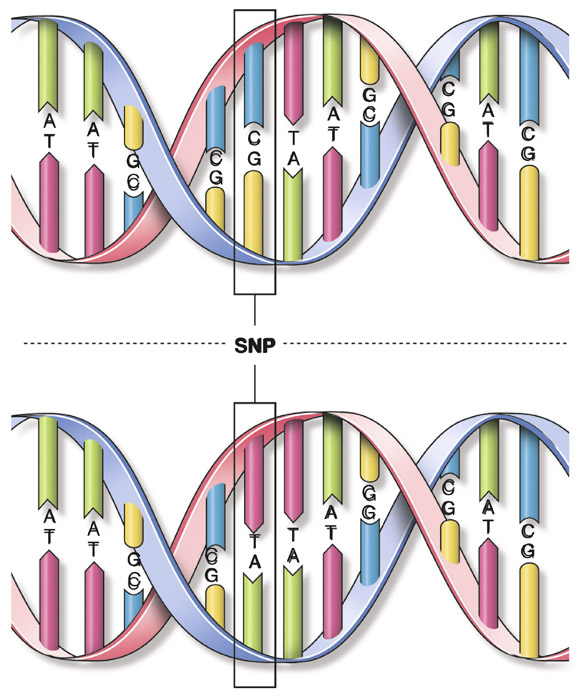
Fig. 4-3 Illustration d’un SNP sous la forme d’un C devenu un T entre la forme de référence (en haut) et la forme mutée (en bas). Dans le cas présent, cette mutation peut provoquer l’apparition d’un codon STOP prématuré (TAG = codon STOP).
Pénétrance : se définit par l’effet clinique de la mutation (une mutation monogénique a une très forte probabilité de se manifester cliniquement) ; une pénétrance faible signifie que la mutation est présente, mais aura un effet clinique modeste ou léger.
Fréquence des allèles : les allèles sont les variations au niveau d’un nucléotide donné, et leur fréquence dans la population peut être forte ou très faible (ce qui est le cas de mutations responsables de formes monogéniques de glaucome : elles sont très rares mais graves).
La première association rapportée dans le glaucome était l’étude sur une cohorte de 195 cas de GPAO islandais comparée à 14 474 témoins. Cette étude a permis de mettre en évidence qu’en fait le gène LOKL1 pouvait représenter une susceptibilité au glaucome par exfoliation et non au GPAO, soulignant l’importance de la classification typique des cas [1, 6, 29].
D’autres travaux importants dans l’historique des études d’association pour le GPAO sont des études japonaises fondées sur le glaucome à pression normale, distinguant les stades évolutifs, caractéristique de ces régions [32].
Le locus de la calvéonine a été le premier locus lié au GPAO par GWAS et pour lequel il y a eu des réplications dans des cohortes indépendantes [29]. Il a été identifié initialement par une étude islandaise (1 263 cas et 34 877 témoins) et répliqué dans des cohortes en Suède, en Grande-Bretagne, en Australie et en Asie. Les SNP associés au locus situé sur le chromosome 7 sont présents dans une zone de déséquilibre de liaison qui contient les gènes CAV1 et CAV2, codant pour la calvéonine 1 et la calvéonine 2. Ces deux protéines sont importantes pour la signalisation cellulaire et l’endocytose, et pourraient influencer la voie du TGF-β et la signalisation monoxyde d’azote (stress oxydant). Des études ont cependant montré un faible effet, et l’association serait plus importante chez les femmes et les personnes ayant un glaucome à pression normale. Les deux gènes codant pour la calvéonine 1 et la calvéonine 2 sont exprimés dans le trabéculum et dans les cellules ganglionnaires de la rétine, et sont impliqués dans la formation des calvéoles qui sont des invaginations de la membrane plasmique fonctionnant comme des vésicules transporteuses macromoléculaires.
Une autre piste est celle du gène TMCOA qui a été identifié comme associé au GPAO en 2011 [3]. Ce gène code pour une protéine qui serait localisée au niveau de l’appareil de Golgi et du réticulum endoplasmique, et qui pourrait avoir un rôle au niveau de l’apoptose des cellules ganglionnaires de la rétine. Il a par ailleurs été identifié dans un syndrome monogénique développemental avec des mutations à l’état homozygote [32]. Dans la même région d’association sont situés trois autres gènes : CDKN2B, ASC1, CDKA2, qui pourraient également être associés.
Des variants dans la région chromosomique 14q23 ont été identifiés comme associés à un CDR de la papille augmenté dans une large étude [26]. Ces variants sont localisés dans des zones intergéniques entre deux gènes : SIX1 et SIX2.
De manière intéressante, les études endophénotypiques semblent montrer un taux de réplication reproduit de façon plus fiable. En effet, trois études indépendantes fondées sur l’ECC ont identifié un locus commun : le locus ZNF469 [18, 26]. Les SNP sont présents sur une zone régulatrice du gène en amont. Ce gène, de manière intéressante quand il est muté, peut conduire au syndrome rare de « brittle cornea » (cornée fragile) [4]. Il existe également une association forte de SNP en plus de ce locus, en tenant compte de l’ECC, pour deux gènes codant pour des formes de collagène COL5A1 et COL8A2 et peut-être pour les gènes FOX01 et AKP13.
La surface de la papille optique a été considérée comme endophénotype dans trois études indépendantes. De manière intéressante, les trois ont identifié le gène ATOH7 et une a pu démontrer une association avec le GPAO [18, 20, 26].
De manière générale, ces études par endophénotype [4] ont permis d’identifier plus précisément un certain nombre de gènes candidats et de locus pour le GPAO.
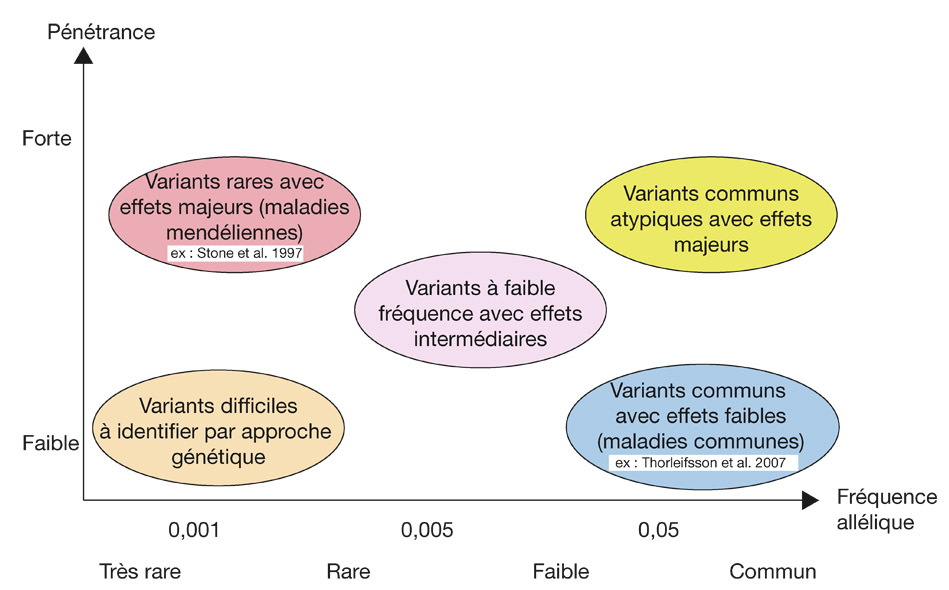
Fig. 4-4 Représentation schématique des variants de faible fréquence comparés aux mutations monogéniques. (D’après Manolio et al., 2009 [21].)
La complexité de la physiopathologie du GPAO est illustrée par la difficulté d’identifier les nombreux facteurs génétiques impliqués et doit tenir compte également des différentes populations étudiées. Un certain nombre de données dans la littérature demandent à être confirmées pour que ces gènes soient fermement l’objet d’identifications biologiques ultérieures. Ces études sont capitales pour comprendre la physiopathologie du glaucome de manière générale, et pourraient identifier de nouvelles voies biologiques impliquées dans le déterminisme et la souffrance des cellules ganglionnaires de la rétine. L’évolution des techniques de biologie moléculaire et de génomique va permettre très rapidement d’affiner ces différentes approches, notamment grâce aux techniques de séquençage haut débit, associée à des progrès en bio-informatique dans le domaine de l’analyse haut débit.
Ainsi il n’est pas à douter qu’à l’avenir, ces études génétiques pourront aboutir à un développement d’une médecine personnalisée, tout particulièrement dans le domaine du glaucome, et devraient permettre également d’identifier des populations à risques particuliers. Une fois ces facteurs génétiques déterminés, de nouvelles voies thérapeutiques s’ouvriront et pourront être affinées par des approches de médecine et thérapies personnalisées.
Les études moléculaires à titre diagnostique sont réservées aux rares cas de familles clairement autosomiques dominantes (transmission verticale) permettant d’identifier les personnes porteuses d’une mutation dans un des gènes connus et d’optimiser sa surveillance.
Les études de facteurs de risque de prédisposition relèvent encore de la recherche et ne permettent pas, à ce jour, d’adapter la surveillance et le traitement de la majorité des patients avec un GPAO.
L’évolution de nos connaissances des facteurs génétiques du GPAO est en plein essor, notamment grâce aux progrès des techniques de séquençage haut débit.
L’identification des zones génomiques associées au GPAO est mieux établie. Des variations génétiques existent dans des régions bien déterminées du génome dénommées locus. Elles ont été rapportées dans le GPAO, le glaucome à pression normale et le glaucome exfoliatif.
Des variants géniques ont été identifiés associés au CDR, à l’ECC et à la surface du disque optique. D’autres sont en cours d’identification. Ces découvertes nous orientent vers des diagnostics de participation génétique sur des facteurs du GPAO (CDR, ECC, etc.) L’amélioration de nos connaissances permettra peut-être d’agir à ce niveau génétique sur ces facteurs de risque.
À l’avenir, l’établissement des facteurs génétiques de prédisposition au GPAO interprétés à travers d’autres éléments (ethniques, endophénotype, environnement, etc.) constitueront le socle de la médecine personnalisée et la base de découvertes physiopathogéniques et thérapeutiques.
[1] Abu-Amero KK, Osman EA, Azad MT, et al. Lack of association between LOXL1 gene polymorphisms and primary open angle glaucoma in the Saudi Arabian population. Ophthalmic Genet. 2012 ; 33 : 130-3.
[2] Baratz KH, Tosakulwong N, Ryu E, et al. E2-2 protein and Fuch’s corneal dystrophy. N Engl J Med 2010 ; 363 : 1016-24.
[3] Burdon KP, Macgregor S, Hewitt AW, et al Genome-wide association study identifies susceptibility loci for open angle glaucoma at TMCO1 and CDKN2B-AS1. Nat Genet. 2011 ; 43 : 574-8.
[4] Burdon KP. Genome-wide association studies in the hunt for genes causing primary open-angle glaucoma : a review. Clin Exp Ophtalmol. 2012 ; 40 : 358-63.
[5] Charlesworth J, Kramer PL, Dyer T, et al. The path to open-angle glaucoma gene discovery : endophenotypic status of intraocular pressure, cup-to-disc ration, and central corneal thickness. Invest Ophtalmol Vis Sci. 2010 ; 51 : 3509-14.
[6] Cheng JW, Cheng SW, Ma XY, et al. Myocilin polymorphisms and primary open-angle glaucoma : a systematic review and meta-analysis. PloS One. 2012 ; 7 : e46632.
[7] Chi ZL, Akahori M, Obazawa M, et al. Overexpression of optineurin E50K disrupts Rab8 interaction and leads to a progressive retinal degeneration in mice. Hum Mol Genet. 2010 ; 19 : 2606-15.
[8] Edwards AO, Ritter III R, Abel KJ, et al. Complement factor H polymorphism and age-related macular degeneration. Science. 2005 ; 308 : 421-4.
[9] Fan BJ, Wang DY, Cheng CY, et al. Different WDR36 mutation pattern in Chinese patients with primary open-angle glaucoma. Mol Vis. 2009 ; 15 : 646-53.
[10] Fingert J. Primary open-angle glaucoma genes. Eye. 2011 ; 25 : 587-95.
[11] Fingert JH, Robin AL, Stone JL, et al. Copy number variations on chromosom 12q14 patients with normal tension glaucoma. Hum Mol Genet. 2011 ; 20 : 2482-94.
[12] Gemenetzi M, Yang Y, Lotery AJ. Current concepts on primary open-angle glaucoma genetics : a contribution to disease pathophysiology and future treatment. Eye. 2012 ; 26 : 355-69.
[13] Green CM, Kearns LS, Wu J, et al. How signifiant is a family history of glaucoma ? Experience from the Glaucoma Inheritance Study in Tasmania. Clin Experiment Ophtalmol. 2007 ; 35 : 793-9.
[14] Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increased the risk of age-related degeneration. Science. 2005 ; 308 : 419-21.
[15] Hauser MA, Allingham RR, Linkroum K, et al. Distribution of WDR36 DANN sequence variants in patients with primary open-angle glaucoma. Invest Ophtalmol Vis Sci. 2006 ; 47 : 2542-46.
[16] Hewitt AW, Mackey DA, Craig JE. Myocilin allele-specific glaucoma phenotype database. Hum Mutat. 2008 ; 29 : 207-11.
[17] Klein RJ, Zeiss C, Chew EY, et al. Complement facteur H polymorphisme in age-related macular degeneration. Science. 2005 ; 308 : 385-9.
[18] Kohr CC, Ramdas WD, Vithana EN, et al. Genome-wide association studies in Asians confirm the involvement of ATOH7 and TGFBR3, and further identify CARD10 as a novel locus influencing optic disc area. Hum Mol Genet. 2011 ; 20 : 1864-72.
[19] Liu Y, Allingham RR. Molecular genetics in glaucoma. Exp Eye Res. 2011 ; 93 : 331-9.
[20] Macgregor S, Hewitt AW, Hysi PG, et al. Genome-wide association identifies ATOH7 as a major gene determining human optic disc size. Hum Mol Genet. 2010 ; 19 : 2716-24.
[21] Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009 ; 461 : 747-53.
[22] Miyazawa A, Fuse N, Mengkegale M, et al. Association between primary open-angle glaucoma and WDR36 DNA sequence variants in Japanese. Mol Vis. 2007 ; 13 : 1912-19.
[23] Morton S, Hesson L, Peggie M, Cohen P. Enhanced binding of TBK1 by an optineurin mutant that causes a familial form of primary open angle glaucoma. FEBS Lett. 2008 ; 582 : 997-1002.
[24] Nakano M, Ikeda Y, Taniguchi T, et al. Three susceptible loci associated with primary open-angle glaucoma identified by genome-wide association study in a Japanese population. Proc Natl Acad Sci USA. 2009 ; 106 : 12838-42.
[25] Pasutto F, Matsumoto T, Mardin CY, et al. Heterozygous NTF4 mutations impariring neurotrophin-4 signaling in patients with primary open-angle glaucoma. Am J Hum Genet. 2009 ; 85 : 447-56.
[26] Ramdas WD, van Koolwijk LM, Ikram MK, et al. A genome-wide association study of optic disc parameters. PLoS Genet. 2010 ; 6 : e1000978.
[27] Rezaie T, Child A, Hitchings R, et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science. 2002 ; 295 : 1077-9.
[28] Stone EM, Fingert JH, Alward WL, et al. Identification of a gene that causes primary open angle glaucoma. Science. 1997 ; 275 : 668-70.
[29] Thorleifsson G, Magnusson KP, Sulem P, et al. Common sequence variants in the LOXL1 gene confers susceptibility to exfoliation glaucoma. Science. 2007 ; 317 : 1397-400.
[30] Vithana EN, Nongpiur ME, Venkataram D, et al. Identification of a novel mutation in the NTF4 gene that causes primary open-angle glaucoma in a Chinese population. Mol Vis. 2010 ; 16 : 1640-45.
[31] Wolfs RC, Klaver CC, Ramrattan RS, et al. Genetic risk of primary open-angle glaucoma. Population-based familial aggregation study. Arch Ophtalmol. 1998 ; 116 : 1640-5.
[32] Xin B, Puffenberger EG, Turben S, et al. Homozygous frameshift mutation in TMCO1 causes a syndrome with craniofacial dysmorphism, skeletal anomalies, and mental retardation. Proc Natl Acad Sci USA. 2010 ; 107 : 258-63.