Dégénérescences et dystrophies cornéennes
V. Borderie
Les dégénérescences cornéennes correspondent à des anomalies acquises de la transparence cornéenne secondaires à des dépôts : lipidiques (gérontoxon, kératopathie lipidique), calcaires (kératopathie en bandelette), amyloïdes, de protéines (kératopathie des dysprotéinémies), hématiques (hématocornée), métalliques, médicamenteux ou de collagène (dégénérescence nodulaire de Salzmann). Elles peuvent être asymptomatiques ou entraîner une baisse de la vision.
Les dystrophies cornéennes correspondent aux pathologies génétiques de la cornée se traduisant par des anomalies bilatérales de la transparence cornéenne. Elles sont classées en fonction du gène atteint et des mutations de ce gène. La dystrophie épithéliale la plus fréquente est la dystrophie de Cogan ou map-dot-fingerprint dystrophy. Elle touche la membrane basale épithéliale. La dystrophie de Meesmann correspond à des mutations des gènes des cytokératines 3 et 12 et se traduit par des microkystes intra-épithéliaux. Les mutations du gène de la kérato-épithéline sont à l’origine de dystrophies de la couche de Bowman et de diverses dystrophies stromales (dystrophies granulaires, grillagées, d’Avellino). Elles se traduisent par des dépôts granulaires et/ou amyloïdes. Leur transmission est autosomique dominante. Les dystrophies épithéliales et celles liées au gène de la kérato-épithéline se traduisent habituellement par des érosions cornéennes douloureuses et une baisse de la vision. Parmi les autres dystrophies stromales, la dystrophie cristalline de Schnyder, la dystrophie gélatineuse et la dystrophie maculaire sont bien caractérisées sur le plan génétique.
Il s’agit d’une infiltration lipidique du stroma cornéen périphérique. Le gérontoxon est constitué de cholestérol, de phospholipides et de triglycérides qui s’accumulent sous la couche de Bowman puis dans la zone prédescemétique. Très fréquent chez les personnes âgées, il peut être également lié à une dyslipidémie chez des patients plus jeunes.
Le gérontoxon n’a pas de retentissement fonctionnel. Au biomicroscope, c’est une mince bande opaque périphérique blanc grisâtre parallèle au limbe, séparée de celui-ci par l’anneau lucide de Vogt (fig. 9-1). En OCT (optical coherence tomography), il est modérément hyperréflectif, mal limité et séparé du limbe par un intervalle de cornée normale (fig. 9-2). En microscopie confocale, on observe des hyperréflectivités dans la couche de Bowman et dans le stroma, ayant un aspect spiculé et organisées en rayons de roue ou en lobules [1].
Le gérontoxon ne requiert aucun traitement. S’il apparaît avant 50 ans, il faut rechercher une hyperlipidémie.
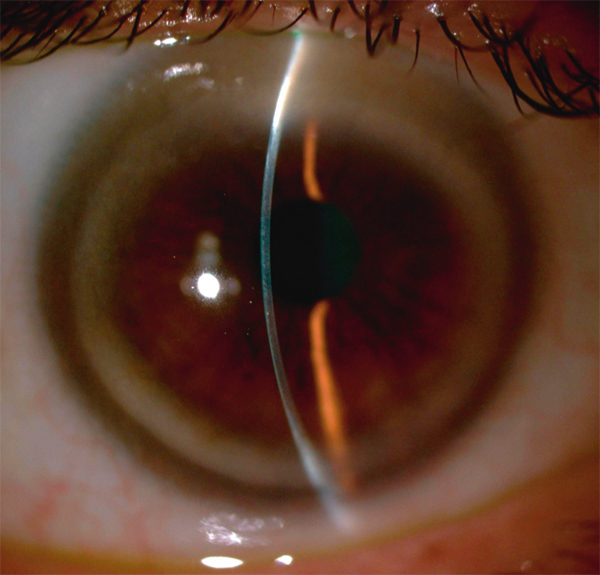
Fig. 9-1 Gérontoxon, aspect biomicroscopique.
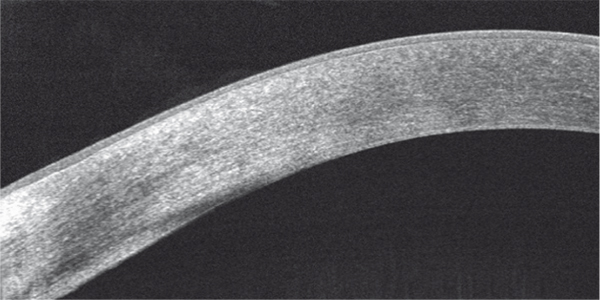
Fig. 9-2 Aspect en OCT d’un gérontoxon.
Discrète hyperréflectivité stromale superficielle périphérique.
Anomalie fréquente (60 % après 40 ans), symétrique, elle se présente sous forme de deux lignes arciformes blanchâtres, temporale et nasale, superficielles et dentelées. Elle correspond à une fragmentation localisée de la périphérie de la couche de Bowman avec une fine surcharge calcaire.
La kératopathie en bandelette correspond à des dépôts calcaires dans la couche de Bowman puis dans le stroma antérieur, typiquement dans l’aire de la fente palpébrale. Elle est habituellement associée à une inflammation chronique de la surface oculaire [2].
Elles peuvent être d’origine oculaire ou générale :
causes oculaires : kératopathie ancienne, traumatismes, cause iatrogène (chirurgie), leucome, dégénérescence sphéroïde, hypotonie, phtyse ;
causes générales : maladie de Still, polyarthrite rhumatoïde, sarcoïdose, ichtyose, hypercalcémie, hyperparathyroïdie, hémodialyse, hypophosphatémie, hypervitaminose D, syndrome de Fanconi, syndrome de Burnett.
Elle se présente sous forme d’une plaque blanchâtre ou grisâtre très fine perforée de multiples orifices, débutant au limbe (fig. 9-3). Elle progresse vers le centre de la cornée et peut donner des érosions douloureuses. Les dépôts siègent typiquement dans l’aire de la fente palpébrale mais la topographie peut différer notamment en cas de forme secondaire à une pathologie cornéenne chronique. En OCT, les dépôts sont hyperréflectifs et siègent dans la couche de Bowman puis progressent dans le stroma antérieur (fig. 9-4). En microscopie confocale, les dépôts sont hyperréflectifs, punctiformes et/ou en mottes [1].
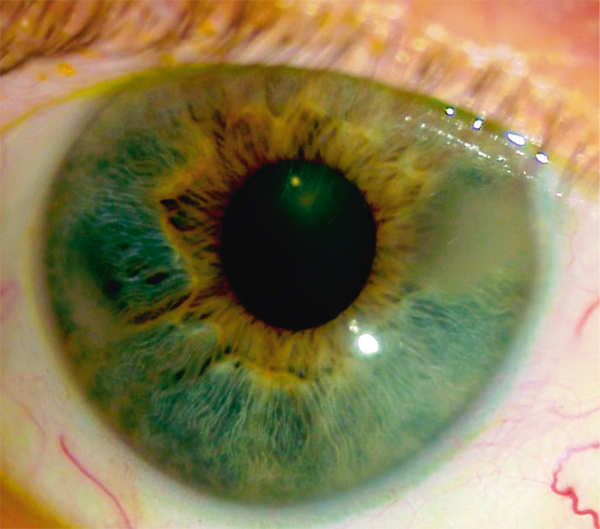
Fig. 9-3 Kératopathie en bandelette, aspect biomicroscopique.
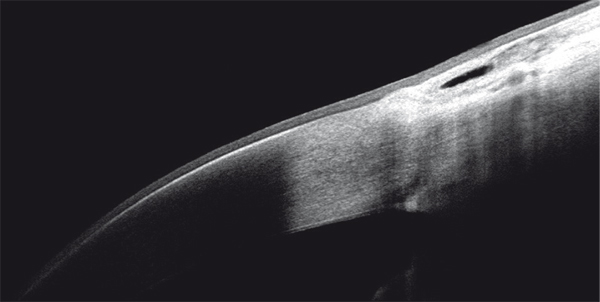
Fig. 9-4 Aspect en OCT d’une kératopathie en bandelette.
Hyperréflectivité dense sous-épithéliale.
On observe des dépôts de grains de calcaire (carbonate ou phosphate de calcium) formant une fine couche au niveau de la couche de Bowman et/ou du stroma superficiel.
Il consiste à faire un pelage de la couche de Bowman et des dépôts sous-jacents. La photokératectomie thérapeutique peut être utilisée et le pelage peut être complété par l’application d’un chélateur, l’EDTA (acide éthylène-diamine-tétra-acétique). La cicatrisation épithéliale postopératoire est parfois longue et une greffe de membrane amniotique peut être utile dans certains cas. Une kératoplastie lamellaire antérieure est parfois nécessaire dans les formes très sévères et/ou récidivantes.
Elle se rencontre notamment après infection herpétique ou zostérienne, mais toute pathologie cornéenne néovascularisée peut entraîner la formation de dépôts lipidiques stromaux. C’est une surcharge stromale jaunâtre dense, d’aspect cristallin, au niveau d’une cicatrice ancienne, autour d’un néovaisseau cornéen stromal (fig. 9-5). Elle est souvent associée à une infiltration calcaire. En OCT, on observe une hyperréflectivité stromale et une augmentation de l’épaisseur cornéenne dans la zone pathologique [1]. Le traitement repose sur le traitement de la pathologie initiale et sur le traitement anti-angiogénique. Ce dernier n’est pas encore codifié [3]. Il peut faire appel aux collyres corticoïdes, à la ciclosporine topique, aux anti-VEGF (vascular endothelial cell growth factor) en injections sous-conjonctivales ou au GS-101 topique.
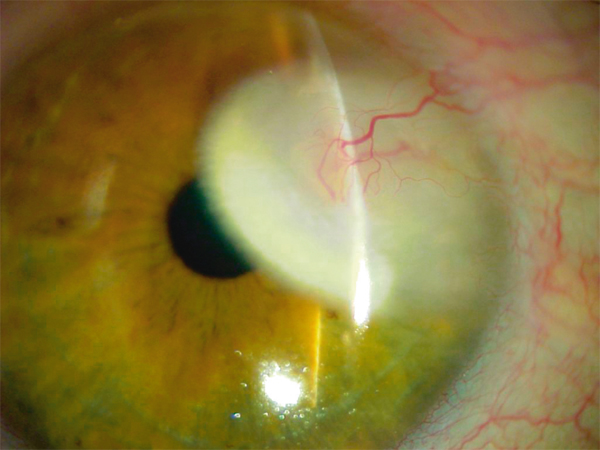
Fig. 9-5 Kératopathie lipidique, aspect biomicroscopique.
C’est une infiltration blanc jaunâtre, granuleuse et fine, sans anneau lucide, souvent supérieure, bilatérale. Il faut rechercher une hyperlipidémie.
Elle se constitue souvent sur une cornée cicatricielle ou après une inflammation chronique, mais semble parfois idiopathique. En histologie, on observe des plaques denses de collagène situées entre l’épithélium et la couche de Bowman avec des dépôts hyalins, des anomalies de la membrane basale épithéliale, de la couche de Bowman et de l’épithélium. Cliniquement, il s’agit de nodules sous-épithéliaux, saillants, opalescents, disposés en forme de couronne autour du centre de la cornée (fig. 9-6). Une fine néovascularisation peut être présente. L’atteinte est uni- ou bilatérale. En OCT, les nodules sont hyperréflectifs et peuvent effacer la couche de Bowman avec une atrophie de l’épithélium (fig. 9-7) [1, 4]. L’épaisseur cornéenne est augmentée au niveau des nodules. En microscopie confocale, les lésions sont des plaques d’hyperréflectivité sous-épithéliales, aspécifiques multiples, régulières, d’aspect fibreux, avec une activation kératocytaire [5]. La prise en charge thérapeutique comporte le traitement des affections inflammatoires associées et les lubrifiants pour les lésions symptomatiques débutantes. Une kératectomie superficielle avec ou sans mitomycine ou une photokératectomie thérapeutique sont indiquées quand la lésion touche l’axe visuel, voire une kératoplastie lamellaire antérieure ou transfixiante. Des récurrences sont possibles après la greffe.
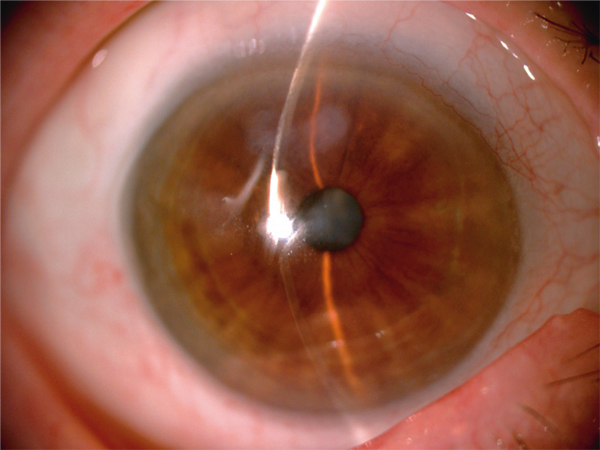
Fig. 9-6 Dégénérescence nodulaire de Salzmann, aspect biomicroscopique.
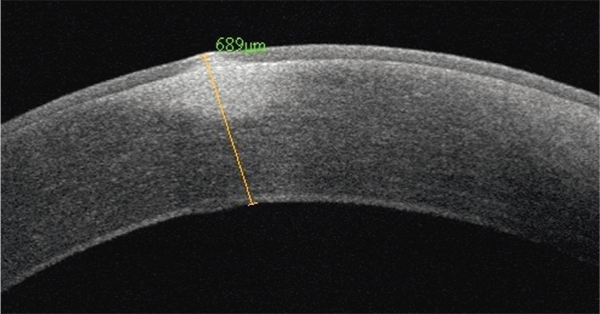
Fig. 9-7 Aspect en OCT d’une dégénérescence nodulaire de Salzmann.
Le nodule est hyperréflectif et efface la couche de Bowman.
Le nom de cette dégénérescence stromale provient de l’aspect de la peau de crocodile. Elle est liée à l’âge mais peut aussi s’observer après certains traumatismes ou en cas de phtyse.
Les lésions biomicroscopiques sont des opacités grisâtres polygonales séparées d’espaces clairs (fig. 9-8), le plus souvent bilatérales et centrales. En OCT, on observe une zone hyperréflective stromale prédominant au centre de la cornée (fig. 9-9). En microscopie confocale, des zones hyperréflectives ponctuées sont présentes dans le stroma antérieur, alors qu’au niveau du stroma profond des lacunes hyporéflectives acellulaires en bandes sont présentes. Des opacités stromales filamenteuses obliques sont visibles, de taille croissante lorsque l’on progresse vers le stroma postérieur [6].
La dégénérescence en crocodile shagreen est asymptomatique et ne requiert aucun traitement.
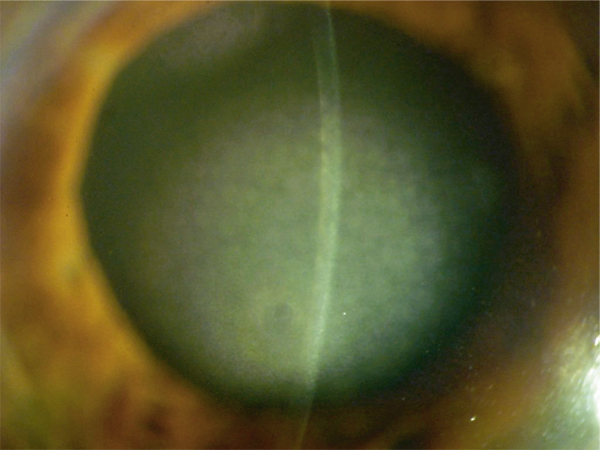
Fig. 9-8 Crocodile shagreen, aspect biomicroscopique.
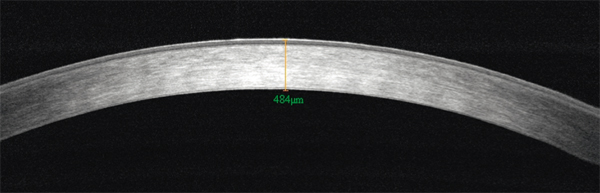
Fig. 9-9 Aspect en OCT d’une dégénérescence cornéenne en crocodile shagreen.
Hyperréflectivité stromale prédominant au centre de la cornée.
Elle se voit chez des patients exposés au soleil, aux ultraviolets, au vent, au sable ou au froid. Il s’agit de nodules translucides puis grisâtres ou bruns dorés, sous-épithéliaux, confluents, bilatéraux, situés dans l’aire de la fente palpébrale, débutant au limbe puis progressant vers le centre de la cornée.
Elle se constitue souvent sur une cornée cicatricielle ou après une inflammation chronique. Il s’agit d’infiltrats gris, blancs ou jaune-orangé sous-épithéliaux. La forme des dépôts est variable : haze stromal, nodules ou plaques sous-épithéliales. En histologie, les dépôts sont colorés par le rouge Congo et la thioflavine T ; ils sont fluorescents en lumière polarisée. Ils ont un aspect microfilamentaire en microscopie électronique à transmission. Une greffe est indiquée en cas de baisse de vision importante.
Cette dégénérescence complique la goutte. Elle se présente comme une opacité jaune sous-épithéliale progressant du limbe vers le centre de la cornée, composée de cristaux d’urate de sodium.
Ces kératopathies se rencontrent dans les cryoglobulinémies, les myélomes, les gammapathies monoclonales bénignes et les lymphomes. Elles se présentent sous forme de dépôts cristallins bilatéraux siégeant au sein de plaques ou d’îlots amorphes blanc grisâtre, souvent dans le stroma moyen mais pouvant intéresser toutes les couches du stroma. Les dépôts ont une topographie périphérique ou périphérique moyenne respectant une zone prélimbique ou centrale. Il n’y a pas ou peu de retentissement fonctionnel. L’atteinte cornéenne doit être distinguée de la dystrophie cristalline [7]. En histologie, il s’agit de dépôts d’immunoglobulines de même nature que l’immunoglobuline monoclonale présente dans le sang. En microscopie électronique à transmission, les dépôts sont extracellulaires, situés dans le stroma, d’architecture cristalline géométrique et périodique (10 nm). Notons qu’une cornea verticillata est également possible au cours du myélome.
Il s’agit d’une imprégnation hématique du stroma cornéen secondaire à un hyphéma associé à une hypertonie. L’opacification du stroma est habituellement irréversible (fig. 9-10).
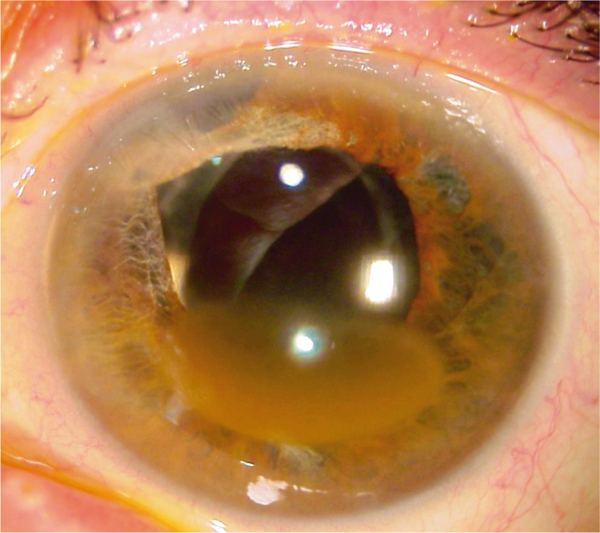
Fig. 9-10 Hématocornée, aspect biomicroscopique.
La sidérose cornéenne est une complication des corps étrangers cornéens ferriques ou d’une sidérose oculaire sévère. Des pigments de ferritine peuvent se déposer physiologiquement en ligne horizontale (ligne de Hudson-Stahli), autour du sommet d’un kératocône (anneau de Fleischer, fig. 9-11), en dedans de l’insertion de la tête d’un ptérygion (ligne de Stocker) et en dedans de l’empiétement cornéen d’une bulle de filtration (ligne de Ferry). En histologie, les dépôts sont situés dans les cellules épithéliales. La chalcose cornéenne se rencontre au cours de la maladie de Wilson (anneau verdâtre limbique de Kayser-Fleisher, à la face postérieure de la cornée) et après corps étranger intra-oculaire en cuivre. La chryséose cornéenne complique les traitements par sels d’or. L’argyrose cornéenne complique un traitement prolongé par collyre au nitrate d’argent.

Fig. 9-11 Anneau de Fleischer, aspect biomicroscopique.
Plusieurs classes de molécules peuvent donner des dépôts cornéens :
l’adrénaline et ses dérivés (accumulation de granules denses entre l’épithélium et la couche de Bowman et dans le stroma antérieur) ;
la chloroquine, la chlorpromazine, l’amiodarone, l’indométacine (surcharge stromale d’aspect stellaire verticillé – cornea verticillata), réversible ;
les sels d’or ;
les fluoroquinolones.
On l’observe au voisinage du limbe, sur une zone cicatricielle, chez les patients mélanodermes, au cours de certaines surcharges (chlorpromazine) et dans la mélanose conjonctivale précancéreuse de Reese.
Une dispersion de pigment mélanique spontanée banale ou excessive (faisceau de Krükenberg au cours du glaucome ; dispersion pigmentaire secondaire à une iridocyclite, un implant de chambre antérieure, une greffe de cornée ou une dystrophie de Fuchs) correspond à la phagocytose des grains de mélanine par les cellules endothéliales.
Les dystrophies cornéennes sont des kératopathies héréditaires bilatérales, avasculaires entraînant une perte de la transparence ou des propriétés optiques de la cornée. Le plus souvent isolées, elles sont parfois associées à un désordre métabolique général. Elles sont liées à une mutation d’un gène [8]. Ceci exclut toutes les dégénérescences cornéennes dont l’origine n’est pas génétique. Néanmoins, sur le plan clinique, la présentation peut être sporadique, un seul membre de la famille étant atteint. Ceci traduit la variabilité d’expression de ces maladies génétiques. La génétique moléculaire permet un diagnostic étiologique précis des dystrophies cornéennes. Auparavant, le diagnostic précis ne pouvait être affirmé qu’en confrontant les données cliniques aux résultats de l’étude histologique et ultrastructurale de la cornée prélevée lors de la greffe. Aujourd’hui, la confrontation entre les données cliniques, les explorations complémentaires et les résultats de la recherche de mutations permet un diagnostic précis dans un grand nombre de cas. Malheureusement, ce diagnostic génétique moléculaire n’est pas un examen de routine et il n’est réalisable que dans peu de centres. Le conseil génétique ne s’est pas encore développé significativement en matière de cornée. Il est pourtant possible devant certaines dystrophies bien caractérisées de déterminer qui est porteur du gène dans la famille. Pour autant être porteur du gène ne signifie pas forcément que l’on va développer la maladie. Les techniques d’imagerie de la cornée (OCT spectral domain et microscopie confocale) sont également très utiles au diagnostic de ces dystrophies en apportant des informations proches de celles obtenues après examen histologique de la cornée prélevée au cours d’une greffe, de manière non invasive et indolore. Elles font actuellement partie intégrante du bilan de ces pathologies cornéennes.
Les progrès de la génétique moléculaire nous ont obligés à reconsidérer la classification des dystrophies cornéennes. Il est actuellement difficile de classer anatomiquement les dystrophies cornéennes opaques en dystrophies épithéliales, sous-épithéliales, stromales et endothélio-descemétiques. Il est logique, aujourd’hui, de les classer en fonction du gène muté. Néanmoins, pour un même gène, plusieurs dystrophies sont possibles en fonction de la localisation de la mutation et du type d’acide aminé remplacé. Ces dystrophies liées à un même gène ont une présentation clinique différente et une localisation dans l’épaisseur de la cornée également différente. Sur le plan clinique, il est possible de séparer les dystrophies stromales (dont l’atteinte peut intéresser la couche de Bowman et le stroma, avec parfois une atteinte associée sous-épithéliale ou descemétique), les dystrophies purement épithéliales et les dystrophies endothélio-descemétiques. La classification génétique utilisée ici est celle de la base OMIM (online mendelian inheritance in man). Les dystrophies endothélio-descemétiques, ne correspondant pas à des pathologies de la surface oculaire, ne sont pas abordées dans cet ouvrage.
C’est la plus fréquente des dystrophies épithéliales. Elle a une prédominance féminine. Elle est le plus souvent sporadique non héréditaire, le mode de transmission est autosomique dominant dans les formes héréditaires. La génétique moléculaire de cette dystrophie est encore mal connue, mais des mutations ont été retrouvées sur le gène de la kérato-épithéline [9].
Les lésions sont bilatérales. La dystrophie se manifeste à l’adolescence ou chez l’adulte par des érosions récidivantes avec typiquement des douleurs le matin au réveil.
À l’examen, les opacités épithéliales sont grisâtres, plutôt centrales et bilatérales. En fonction de la forme clinique, ces opacités peuvent être punctiformes (microkystes), arrondies et regroupées en archipel (kystes de grande taille), en carte de géographie ou en forme d’empreintes digitales (fig. 9-12). Ces opacités sont variables et labiles dans le temps.
En OCT, on observe une hyperréflectivité des kystes dans l’épithélium, une membrane basale pluristratifiée dont les différentes lamelles sont hyperréflectives, une hétérogénéité de l’épaisseur épithéliale avec des zones d’épaississement et des zones d’amincissement (fig. 9-13) [1]. En microscopie confocale, les lésions sont hétérogènes : lacunes au sein de la couche des cellules basales épithéliales ; points hyperréflectifs au sein de la couche des cellules basales épithéliales ; grandes plages d’hyperréflectivité ; zones bien limitées hyperréflectives en carte de géographie ; lésions arrondies hyporéflectives entourées de liserés hyperréflectifs au niveau basal épithélial ; éléments de morphologie dendritique de grande taille ; stries hyporéflectives parallèles au sein de l’épithélium ; filets hyperréflectifs s’invaginant dans la couche des cellules basales épithéliales (fig. 9-14) [10].
L’examen histologique montre : des microkystes colorés par le PAS (periodic acid Schiff) intra-épithéliaux ; des cellules épithéliales remaniées œdémateuses ; une pseudo-membrane basale aberrante intra-épithéliale épaissie et déformée formant des invaginations en doigt de gant dans l’épithélium ; une couche de Bowman normale. En microscopie électronique à transmission, on observe des kystes intra-épithéliaux contenant des débris cellulaires et un matériel fibrillaire et granuleux synthétisé par les cellules basales en position normale ou anormale.
En fonction de la symptomatologie fonctionnelle, le traitement pourra faire appel à des collyres mouillants, au port d’une lentille thérapeutique et à la photokératectomie thérapeutique.
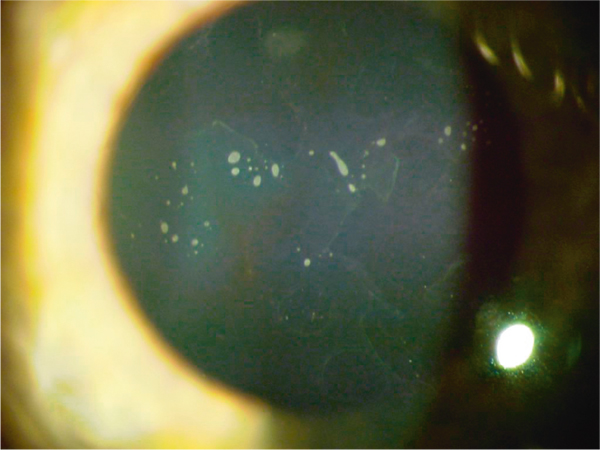
Fig. 9-12 Dystrophie de Cogan vésiculaire, aspect biomicroscopique.
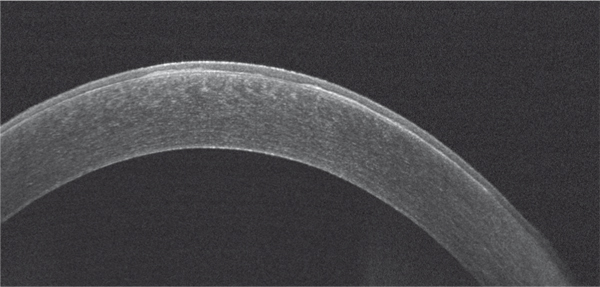
Fig. 9-13 Aspect en OCT d’une dystrophie de Cogan.
Hyperréflectivité et épaississement irrégulier de la membrane basale épithéliale.
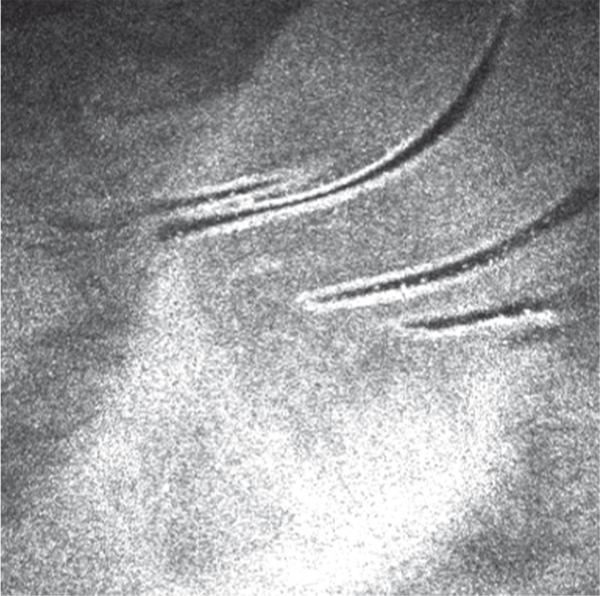
Fig. 9-14 Aspect en microscopie confocale d’une dystrophie de Cogan.
Hyperréflectivité de la membrane basale épithéliale qui est plissée.
La transmission est autosomique dominante avec une pénétrance incomplète. Les mutations sont localisées sur les gènes des cytokératines CK12 (17q12) et CK3 (12q13). La plupart intéressent le gène de CK12 : M129T (CK12), Q130P (CK12), N133K (CK12), R135T (CK12), R135 Gly (CK12), R135 Ile (CK12), L140R (CK12), V143L (CK12), T429D (CK12) [4, 11]. L’expression de la paire de cytokératines 3 et 12 (filaments intermédiaires du cytosquelette des cellules épithéliales) est un marqueur de différenciation cornéenne de l’épithélium de la surface oculaire.
L’atteinte est bilatérale et symétrique. La dystrophie se manifeste à l’adolescence ou chez l’adulte jeune, parfois dans la petite enfance (vers 1–2 ans), par des crises douloureuses (érosions épithéliales récidivantes) avec photophobie et larmoiement. Elle peut retentir sur la vision à un stade évolué. L’examen met en évidence des kystes intra-épithéliaux punctiformes (vésicules) dont le nombre augmente avec l’âge (fig. 9-15), une opacification sous-épithéliale tardive. Les lésions sont diffuses.
En OCT, l’épaisseur épithéliale est irrégulière, l’épithélium est globalement hyperréflectif, surtout en superficie, avec de fines ponctuations hyperréflectives (fig. 9-16). En microscopie confocale, les microkystes se traduisent par des zones aréflectives dépourvues de cellules épithéliales avec des débris cellulaires au sein de l’épithélium (fig. 9-17) [12].
Le diagnostic histologique peut être fait à partir d’un pelage épithélial : épithélium épaissi ; dyskératose épithéliale ; kystes PAS positifs intra-épithéliaux avec un matériel amorphe intrakystique ; halo clair périnucléaire au niveau de cellules épithéliales ; membrane basale épaissie multilamellaire ; couche de Bowman intacte. En microscopie électronique à transmission, on observe un matériel dense fibrillaire et granuleux dans le cytoplasme des cellules épithéliales surtout basales et dans la lumière des kystes intra-épithéliaux correspondant à des filaments de cytokératine, ainsi qu’une membrane basale épaissie et lamellaire.
Les crises douloureuses sont améliorées par un traitement mouillant ou par le port de lentilles souples. Une photokératectomie thérapeutique peut être proposée, mais la dystrophie récidive après traitement.

Fig. 9-15 Dystrophie de Meesmann, aspect biomicroscopique.
Microkystes intra-épithéliaux visibles en rétro-illumination.
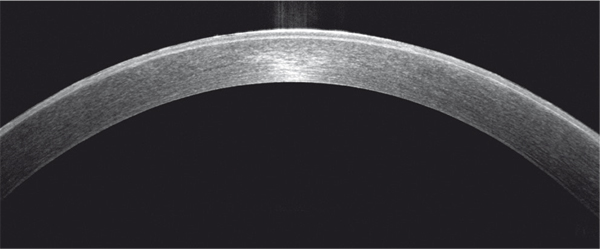
Fig. 9-16 Aspect en OCT d’une dystrophie de Meesmann.
Hyperréflectivité épithéliale.
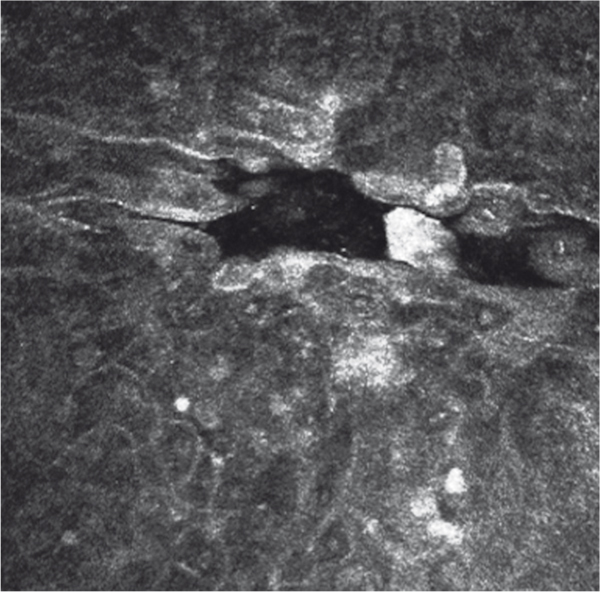
Fig. 9-17 Aspect en microscopie confocale d’une dystrophie de Meesmann. Microkyste intra-épithélial.
La transmission est récessive liée à l’X. Le gène est localisé en Xp22.3 [4]. Les lésions apparaissent dans l’enfance sous forme d’opacités superficielles linéaires, radiaires, respectant le centre de la cornée. L’évolution au cours de la vie se fait vers l’atteinte centrale avec des opacités en forme de flammes. Une photophobie s’installe et s’aggrave avec l’évolution des opacités. Une baisse de vision est possible dans les formes évoluées. En histologie, des microkystes sont présents dans toute l’épaisseur de l’épithélium. En microscopie électronique à transmission, les cellules épithéliales comportent une substance dense homogène ou lamellaire.
La transmission est autosomique dominante avec une prédominance masculine. La dystrophie se manifeste chez l’enfant ou l’adulte par des érosions épithéliales récidivantes, une photophobie intense, parfois une baisse de vision. Les lésions sont centrales uni- ou bilatérales : pertes de substance ; bulles et kystes épithéliaux ; voile sous-épithélial. En histologie, on observe : des microkystes intra-épithéliaux ; un œdème épithélial focal intra- et extracellulaire ; des défauts de cicatrisation de la membrane basale ; une déficience des hémidesmosomes.
Elle est autosomique dominante. Elle débute chez l’enfant ou l’adolescent par des érosions douloureuses, une photophobie, une baisse de vision liée à l’irrégularité épithéliale. On observe des opacités gris blanchâtre en mottes au niveau de la couche de Bowman se prolongeant dans l’épithélium, centrales et bilatérales, avec une membrane basale épaissie. En histologie, on observe : des dépôts PAS positifs au niveau de la membrane basale à extension intra-épithéliale ; une dégénérescence des cellules basales ; l’absence focale de couche de Bowman. Certains considèrent cette dystrophie comme une variante de la dystrophie de Reis-Bücklers [13].
Elle est récessive liée à l’X (maladie de Fabry, déficit en céramide α-galatosidase A) et apparaît chez le sujet jeune.
Il n’y a pas de signes fonctionnels. L’examen montre des lignes brunes arquées ou spiralées, en forme de comète centrée par un point situé dans la partie inférieure de la cornée, et des angiokératomes cutanés diffus (fig. 9-18). On observe : en OCT, une discrète hyperréflectivité épithéliale ; en microscopie confocale, des inclusions hyperréflectives intracellulaires dans la couche basale de l’épithélium cornéen et dans l’épithélium conjonctival. Ces anomalies précèdent l’apparition des signes cliniques [14, 15].
En histologie, on note : des dépôts de glycolipides (à structure lamellaire, très osmiophiles, en microscopie électronique) dans les cellules épithéliales basales ; une membrane basale irrégulière dédoublée, la couche de Bowman parfois atteinte.

Fig. 9-18 Cornea verticillata, aspect biomicroscopique.
Le syndrome KID (keratitis, ichtyosis, deafness) est rare et se transmet sur le mode autosomique dominant. Il est lié à des mutations du gène de la connexine 26, protéine exprimée par les gap junctions de l’épithélium cornéen, de l’épiderme et des annexes. Les formes familiales sont rares et la présentation est le plus souvent sporadique.
Cliniquement, le syndrome associe une kératite (95 %), une insuffisance limbique avec néovascularisation cornéenne dont l’apparition peut être retardée (80 %), une surdité de perception (90 %), une érythrokératodermie donnant un aspect rugueux à la peau (90 %), une alopécie (80 %), une hyperkératose palmoplantaire réticulée (40 %). À un stade précoce, l’atteinte oculaire peut se limiter à des ulcérations cornéennes, une photophobie et des conjonctivites. Une dyskératose de l’épithélium cornéen et conjonctival a été décrite.
Les dystrophies cornéennes dont la génétique moléculaire est la mieux connue sont celles qui sont liées à des mutations du gène βIGH3 (TGF-β-induced gene localisé en 5q31, 2691 paires de bases) codant pour la kérato-épithéline, protéine ubiquitaire d’un poids moléculaire de 68 kD, faite de 683 acides aminés avec un motif RGD à l’extrémité carboxylée, qui se lie aux collagènes I, II, IV et VI [4]. Ces dystrophies atteignent le stroma cornéen depuis la couche de Bowman jusqu’au stroma postérieur. La kérato-épithéline est sécrétée majoritairement par l’épithélium cornéen. Elle est présente dans la cornée normale au niveau de l’épithélium limbique et cornéen, entre les cellules, au niveau de la couche de Bowman et du stroma. Elle est un des principaux composants des dépôts retrouvés au sein des cornées dystrophiques. Les dystrophies liées à des mutations de la kérato-épithéline donnent soit des dépôts granulaires, soit des dépôts amyloïdes, soit des dépôts mixtes. Deux grands mécanismes peuvent expliquer la formation d’amylose : les mutations de la protéine précurseur et la protéolyse. Toutes les dystrophies liées à des mutations du gène de la kérato-épithéline récidivent après greffe. Le tableau 9-1 présente des caractéristiques de ces diverses dystrophies.
Les autres dystrophies stromales non liées au gène de la kérato-épithéline sont moins fréquentes. Les principales sont présentées dans le tableau 9-2. Il faut noter que parmi les dystrophies amyloïdes de la cornée, certaines sont liées au gène de la kérato-épithéline (dystrophies grillagées de type I et III) et d’autres sont liées à d’autres gènes (dystrophie grillagée de type II, dystrophie gélatineuse).
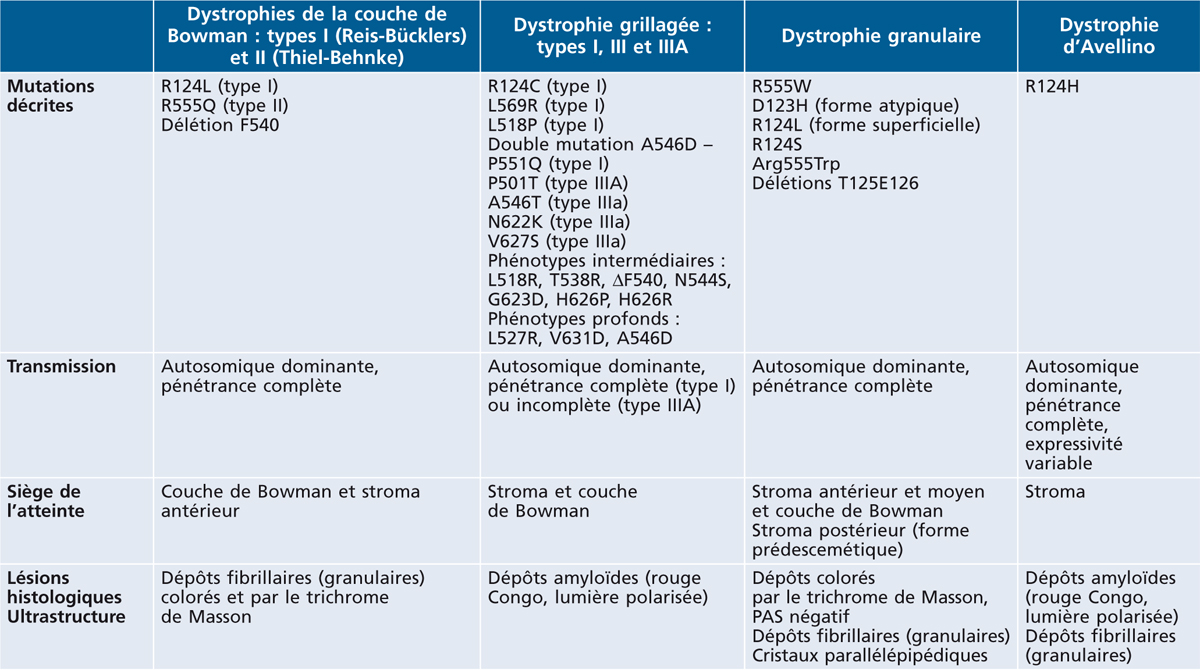
Tableau 9-1 Dystrophies stromales liées à des mutations du gène de la kérato-épithéline.
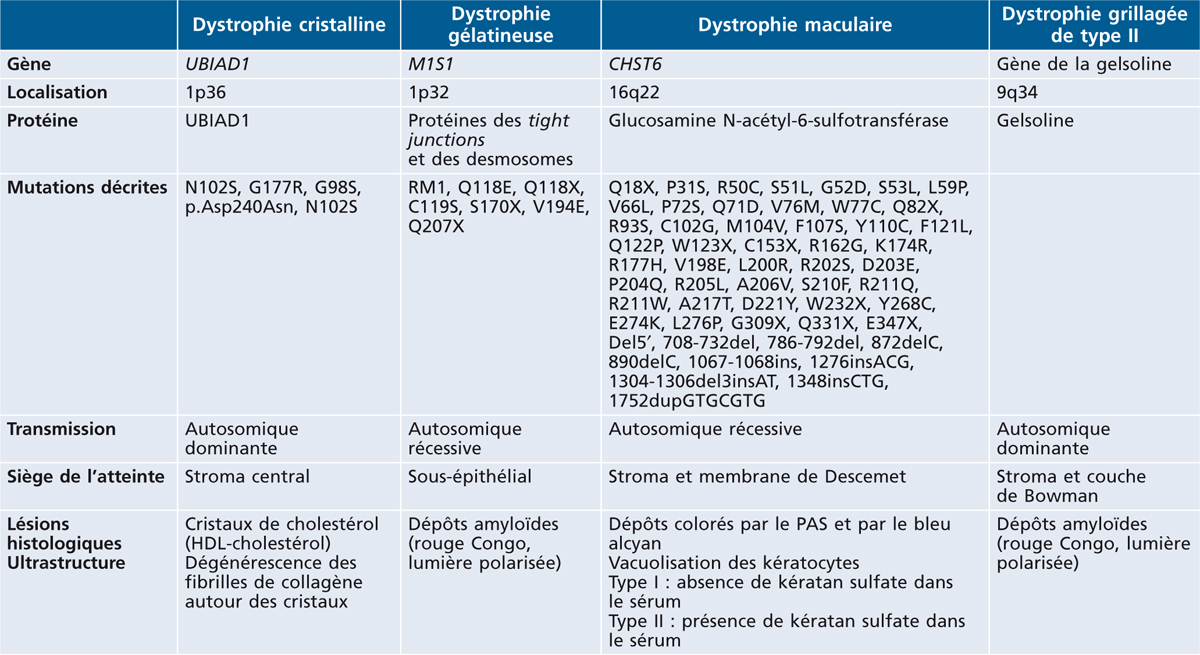
Tableau 9-2 Principales dystrophies stromales non liées au gène de la kérato-épithéline.
Cette dystrophie se transmet sur le mode autosomique dominant ; elle appartient au groupe de dystrophies liées au gène de la kérato-épithéline. Elle se déclare dans l’enfance. Elle se manifeste par des érosions épithéliales récidivantes, diminuant en fréquence après 30 ans. La baisse d’acuité visuelle est progressive et devient importante vers 20–30 ans.
Les opacités sont bilatérales et symétriques, confluentes, à contours géographiques, situées dans le plan de la couche de Bowman au niveau de la cornée centrale, avec un aspect vitreux ou granuleux du stroma antérieur en rétro-illumination (fig. 9-19). Il s’agit d’une forme superficielle de dystrophie granulaire. Elle est également dénommée dystrophie granulaire type III. On observe : en OCT, une plage hyperréflective sous-épithéliale ; en microscopie confocale, des petits dépôts très réflectifs sans zone d’ombre (fig. 9-20) [1]. Les dépôts sous-épithéliaux sont plus réflectifs dans la dystrophie de Reis-Bücklers que dans celle de Thiel-Behnke [16].
Les lésions histologiques sont un épithélium épaissi et irrégulier avec des prolongements dans le stroma antérieur, des disparitions localisées de la couche de Bowman, des dépôts granulaires situés au niveau de la couche de Bowman et du stroma antérieur. Ces dépôts sont colorés par le trichrome de Masson. Ils correspondent à la kérato-épithéline. En microscopie électronique à transmission, ils ont une forme de bâtonnets.
Le traitement repose sur la photokératectomie thérapeutique puis la greffe lamellaire antérieure quand la baisse de vision est importante. La dystrophie récidive de manière fréquente et précoce après kératoplastie.
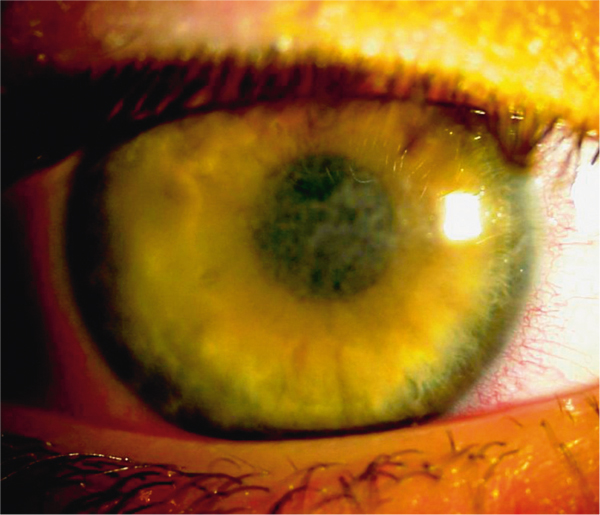
Fig. 9-19 Dystrophie de Reis-Bücklers, aspect biomicroscopique.
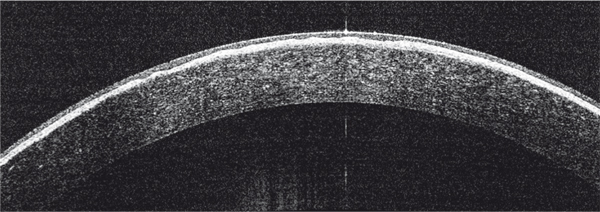
Fig. 9-20 Aspect en OCT d’une dystrophie de Reis-Bücklers.
Hyperréflectivité dense sous-épithéliale irrégulière.
Cette dystrophie se transmet sur le mode autosomique dominant ; elle appartient au groupe de dystrophies liées au gène de la kérato-épithéline. Elle se déclare dans l’enfance et se manifeste par des érosions épithéliales récidivantes. Une baisse d’acuité visuelle est possible, mais plus tardive que dans la dystrophie de Reis-Bücklers. La progression est lente. La baisse d’acuité visuelle devient importante vers la cinquantaine.
L’examen montre des opacités blanchâtres centrales sous-épithéliales bilatérales en rayons de miel (fig. 9-21). La présentation clinique est parfois difficile à différencier de celle de la dystrophie de Reis-Bücklers. L’aspect en rayons de miel est plus marqué dans la dystrophie de Thiel-Behnke. En OCT, on observe une plage hyperréflective sous-épithéliale continue, crénelée, en dents de scie [1]. Cette irrégularité est masquée par l’épithélium qui lisse la surface cornéenne (fig. 9-22). En microscopie confocale, les dépôts sont homogènes et arrondis.
Les lésions histologiques sont une destruction de la couche de Bowman avec : un épithélium constitué de cellules de taille et de forme irrégulières ; une membrane basale dédoublée et épaissie ; une disparition de la membrane basale à de nombreux endroits ; un tissu fibrocellulaire sous-épithélial d’aspect ondulé. En microscopie électronique à transmission, on observe une couche fibrillaire et granuleuse sous-épithéliale, avec une perte des hémidesmosomes des cellules épithéliales basales, des fibrilles de collagène courtes arciformes (curly fibers) d’un diamètre de 9 à 15 nm et des dépôts lipidiques dans le stroma [17].
Le traitement repose sur la photokératectomie thérapeutique puis la greffe lamellaire antérieure quand la baisse de vision est importante. La dystrophie récidive après kératoplastie, mais la récidive est plus tardive que dans la dystrophie de Reis-Bücklers.
Fig. 9-21 Dystrophie de Thiel-Behnke, aspect biomicroscopique.
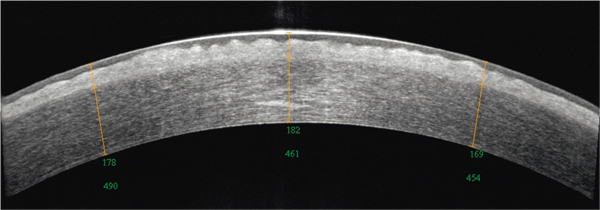
Fig. 9-22 Aspect en OCT d’une dystrophie de Thiel-Behnke.
Hyperréflectivité sous-épithéliale irrégulière, en dents de scie, moins dense que dans la dystrophie de Reis-Bücklers.
Cette dystrophie se transmet sur le mode autosomique dominant ; elle appartient au groupe de dystrophies liées au gène de la kérato-épithéline. Elle est bilatérale et symétrique avec un début précoce dans la petite enfance. Les érosions épithéliales récidivantes constituent la première manifestation clinique. La baisse d’acuité visuelle est progressive. Elle conduit souvent à la greffe.
La forme classique comporte des dépôts granuleux blanchâtres (en flocons de neige ou en mie de pain, diamètre < 300 µ) avec des zones saines au niveau des couches antérieure et moyenne du stroma central (fig. 9-23). La forme diffuse superficielle (homozygote) intéresse toute la surface de la cornée, avec des dépôts au niveau de la couche de Bowman et du stroma antérieur, un aspect en verre de cathédrale ou en voile, des lésions de la couche de Bowman. La forme granuleuse prédescemétique se caractérise par de petits dépôts cristallins blanchâtres postérieurs. En OCT, les dépôts sont hyperréflectifs, bien limités, de forme volontiers polygonale (fig. 9-24) [1]. La surface stromale est irrégulière et ondulée avec un lissage par l’épithélium qui s’hyperplasie dans les zones creuses et s’hypoplasie dans les zones surélevées. En microscopie confocale, les dépôts sont hyperréflectifs et inhomogènes [18].
Les lésions histologiques sont un épithélium irrégulier avec des zones de disparition de la couche de Bowman et des dépôts colorés en rouge par le trichrome de Masson, PAS négatif. Des cristaux parallélépipédiques agglomérés en masse sont visualisés en microscopie électronique à transmission. Les dépôts sont constitués de kérato-épithéline.
Dans les formes superficielles, la photokératectomie thérapeutique est utilisée tant qu’il reste suffisamment d’épaisseur stromale en arrière des dépôts. Lorsque l’atteinte est profonde, le traitement repose sur la kératoplastie lamellaire antérieure profonde qui donne de bons résultats mais la pathologie récidive rapidement après la greffe.
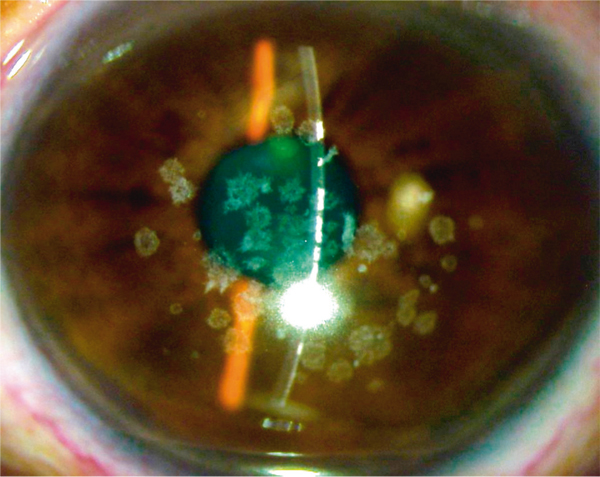
Fig. 9-23 Dystrophie granulaire, aspect biomicroscopique.
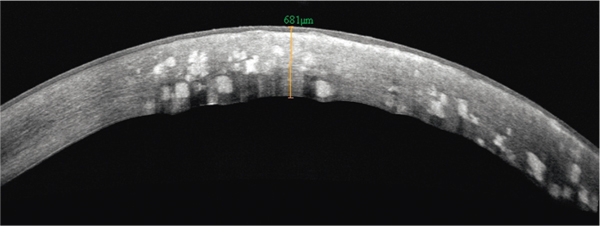
Fig. 9-24 Aspect en OCT d’une dystrophie granulaire.
Dépôts stromaux hyperréflectifs polygonaux bien limités.
Cette dystrophie se transmet sur le mode autosomique dominant ; elle appartient au groupe de dystrophies liées au gène de la kérato-épithéline. La dystrophie se déclare dans l’enfance.
Elle associe des lésions biomicroscopiques et histologiques de dystrophie granulaire à des lésions de dystrophie grillagée volontiers disposées en feux d’artifice (fig. 9-25). Les formes homozygotes sont extrêmement précoces, presque congénitales. Les dépôts peuvent être augmentés par un Lasik (laser in situ keratomileusis) ; la récurrence des dépôts après photokératectomie thérapeutique est plus rapide et plus intense en cas de mutation homozygote R124H qu’en cas de mutation hétérozygote R124H.

Fig. 9-25 Dystrophie d’Avellino, aspect biomicroscopique.
Cette dystrophie se transmet sur le mode autosomique dominant avec une pénétrance complète ; elle appartient au groupe de dystrophies liées au gène de la kérato-épithéline [4]. Parmi les différents types de dystrophies grillagées, le type I est le plus fréquent.
La dystrophie débute chez l’enfant (première ou deuxième décennie). L’atteinte est bilatérale et symétrique. Les signes fonctionnels sont des érosions récidivantes puis une baisse d’acuité visuelle progressive. À la lampe à fente, on observe des dépôts punctiformes superficiels au centre de la cornée puis des lignes cristallines irrégulières se dichotomisant en « crins de Florence » ou en « sucre candi » et une coloration jaunâtre de la cornée, parfois une galette dense disciforme centrale (fig. 9-26). L’évolution se fait vers une extension en profondeur et en périphérie. En OCT, les dépôts sont hyperréflectifs, mal limités, la surface stromale est irrégulière et ondulée (fig. 9-27) [1]. La récidive après greffe débute dans la zone sous-épithéliale. En microscopie confocale, les dépôts sont hyperréflectifs, présents au niveau de ou sous la couche basale épithéliale et dans le stroma avec une augmentation de la réflectivité de la matrice extracellulaire, des images linéaires hyperréflectives typiquement ramifiées et des structures réflectives d’aspect ondulé. Le plexus nerveux sous-épithélial présente des anomalies [19].
En histologie, on observe des dépôts amyloïdes (colorés par le rouge Congo, fluorescents après coloration par la thioflavine T et examen en lumière ultraviolette, biréfringents en lumière polarisée) ovalaires entre les lamelles du stroma et des ruptures de la couche de Bowman. La kérato-épithéline s’accumule au sein des dépôts.
Dans les formes assez superficielles, la photokératectomie thérapeutique est utilisée tant qu’il reste suffisamment d’épaisseur stromale. Dans les atteintes profondes, la kératoplastie lamellaire antérieure profonde donne de bons résultats mais la pathologie récidive après la greffe.
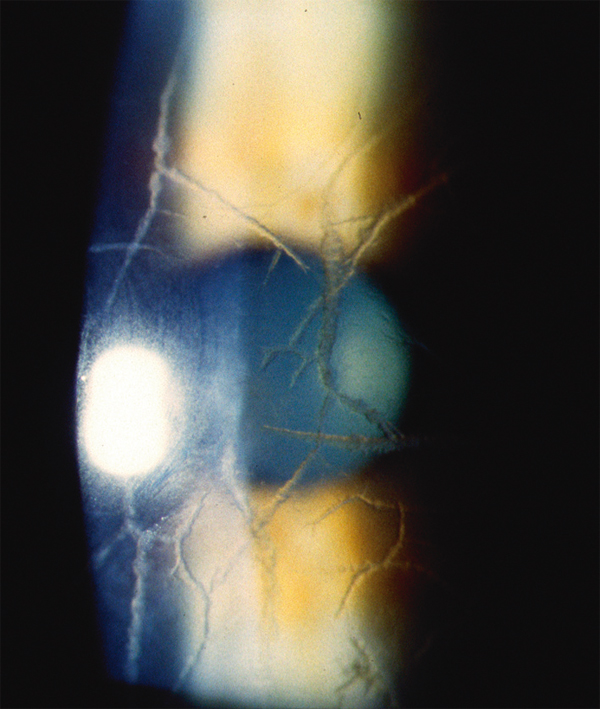
Fig. 9-26 Dystrophie grillagée, aspect biomicroscopique.
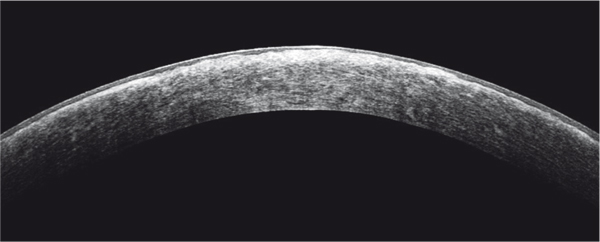
Fig. 9-27 Aspect en OCT d’une dystrophie grillagée.
Dépôts stromaux hyperréflectifs mal limités
Cette dystrophie se transmet sur le mode autosomique dominant avec une pénétrance incomplète ; elle appartient au groupe de dystrophies liées au gène de la kérato-épithéline [4]. Par rapport à la dystrophie grillagée de type I, le type IIIa se caractérise par un début plus tardif vers 40 ans, parfois après 70 ans, des dépôts plus épais s’étendant de limbe à limbe, des opacités nodulaires, puis un voile diffus.
Cette dystrophie se transmet sur le mode autosomique dominant avec une pénétrance complète ; elle appartient au groupe de dystrophies liées au gène de la kérato-épithéline [4]. Par rapport aux dystrophies grillagées de type I et IIIa, le type I/IIIa se caractérise par un début intermédiaire vers 20–40 ans. Elle est souvent asymétrique.
Cette dystrophie se transmet sur le mode autosomique dominant avec une pénétrance complète ; elle appartient au groupe de dystrophies liées au gène de la kérato-épithéline [4]. Par rapport aux dystrophies grillagées de type I, IIIa et I/IIIa, le type III se caractérise par un début vers 40–50 ans, une présentation fortement asymétrique et des lésions inaugurales dans le stroma postérieur. La progression des lésions se fait du stroma postérieur vers le stroma antérieur à l’inverse des autres dystrophies grillagées.
C’est une dystrophie liée au gène de la gelsoline situé sur le chromosome 9q34 à transmission autosomique récessive [4]. Le syndrome de Meretoja comporte des dépôts cornéens grillagés, une neuropathie bilatérale et symétrique des nerfs crâniens et des nerfs périphériques et un lichen amyloïde cutané.
C’est une anomalie du métabolisme du kératan sulfate (défaut de sulfatation du kératan). La transmission est autosomique récessive et le gène atteint est celui d’une carbohydrate sulfotransférase (CHST6) située en 16q21 [4].
Elle se manifeste au cours de l’adolescence, mais les premières lésions peuvent s’observer avant 10 ans. L’atteinte est bilatérale et symétrique. Les signes fonctionnels sont une baisse d’acuité visuelle et une photophobie importantes, ainsi que des érosions cornéennes. À l’examen, les dépôts sont mal limités, confluents, d’aspect blanc sale, occupant toute l’épaisseur du stroma, respectant une zone prélimbique, avec un haze entre les dépôts (fig. 9-28). L’atteinte est superficielle au départ puis progresse vers le stroma postérieur. L’épaisseur cornéenne est diminuée. La sensibilité cornéenne diminue au cours de l’évolution. Les lésions sont souvent associées à une atteinte des cellules endothéliales et une cornea guttata. En OCT, les dépôts donnent une hyperréflectivité stromale mal délimitée avec une surface stromale irrégulière et ondulée (fig. 9-29) [1]. En microscopie confocale, ils sont hyperréflectifs avec un aspect strié [20].
En histologie, les dépôts sont colorés par le PAS (glycosaminoglycanes) et le bleu alcyan ; ils sont situés dans le stroma, les kératocytes, la membrane de Descemet et les cellules endothéliales. Les kératocytes présentent de nombreuses vacuoles en microscopie électronique.
Très lentement évolutive, la dystrophie maculaire conduit habituellement à la kératoplastie transfixiante après l’âge de 30–40 ans. Elle ne récidive pas sur le greffon. La kératoplastie lamellaire antérieure profonde ne donne pas de bons résultats dans la dystrophie maculaire à cause de l’atteinte endothélio-descemétique [21].
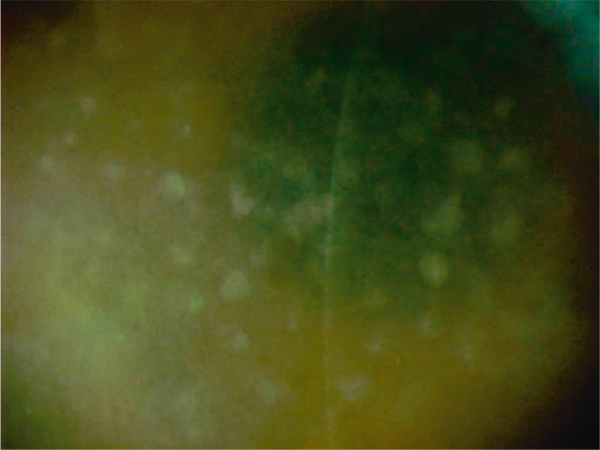
Fig. 9-28 Dystrophie maculaire, aspect biomicroscopique.
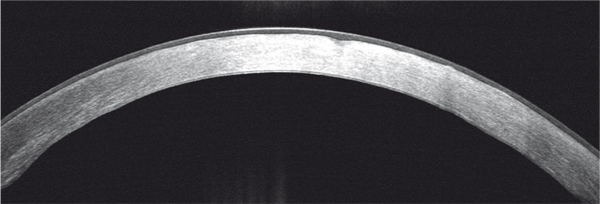
Fig. 9-29 Aspect en OCT d’une dystrophie maculaire.
Hyperréflectivité stromale mal délimitée.
Il s’agit de dépôts de cristaux de cholestérol et de phospholipides dans le stroma cornéen associés à une dégénérescence des fibrilles de collagène. La transmission est autosomique dominante, le gène en cause est UBIAD1 situé sur le chromosome 1p36.3. Le début peut se faire pendant la petite enfance avec une baisse de vision progressive et une hypercholestérolémie familiale associée dans deux tiers des cas.
L’examen montre une atteinte bilatérale et symétrique avec un haze et/ou une accumulation de cristaux irisés (absents dans la moitié des cas) dans la portion centrale du stroma (fig. 9-30). Au cours de l’évolution, une hypoesthésie cornéenne peut apparaître. L’atteinte est centrale au départ puis progresse avec un arc lipidique périphérique puis un haze en moyenne périphérie. En OCT, on observe une hyperréflectivité diffuse du stroma, maximale sous l’épithélium (fig. 9-31) [1]. En microscopie confocale, les dépôts stromaux sont hyperréflectifs avec un aspect filiforme et une organisation en réseau cristallin dans le stroma antérieur ou en lignes parallèles (fig. 9-32) [22].
En histologie, on note des cristaux de cholestérol dans le stroma (HDL-cholestérol en immunohistochimie) avec une dégénérescence des fibrilles de collagène autour des cristaux en microscopie électronique à transmission.
Le traitement repose habituellement sur la kératoplastie lamellaire ou transfixiante après l’âge de 30–40 ans. Une photokératectomie thérapeutique pet être utile en cas de dépôts très superficiels.
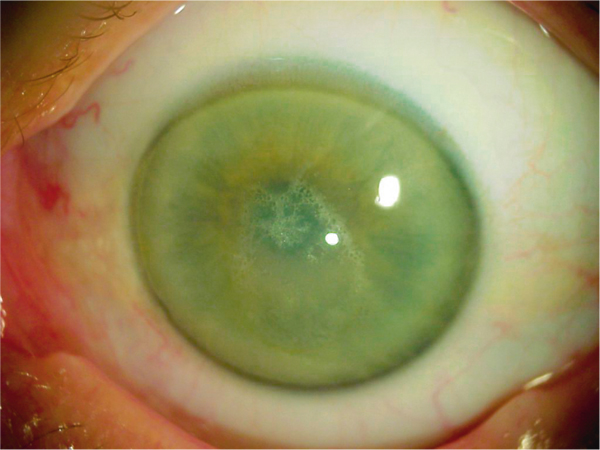
Fig. 9-30 Dystrophie cristalline, aspect biomicroscopique.
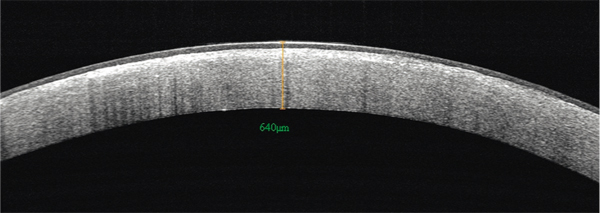
Fig. 9-31 Aspect en OCT d’une dystrophie cristalline.
Hyperréflectivité stromale diffuse maximale sous l’épithélium.
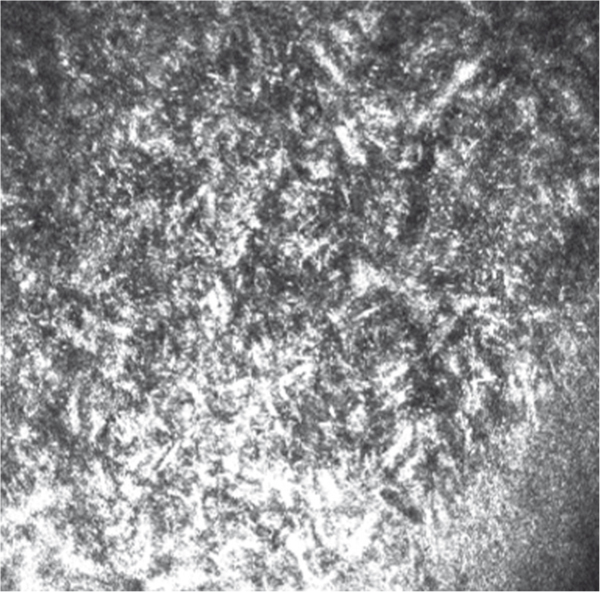
Fig. 9-32 Aspect en microscopie confocale d’une dystrophie cristalline.
Dépôts hyperréflectifs de morphologie cristalline.
Elle est autosomique dominante, à début tardif (forme sénile) ou dans l’enfance. Il n’y a pas de signes fonctionnels ou bien une baisse de vision minime. Les opacités gris blanchâtre sous-épithéliales sont polygonales en écailles (grains de calcaire) séparées par des espaces clairs, avec lésions centrales bilatérales de la couche de Bowman. En histologie, on note des dépôts calcaires au niveau de la couche de Bowman et du stroma antérieur.
Elle est autosomique dominante ou bien récessive liée à l’X. Le début est congénital, dans l’enfance ou chez l’adulte. Une baisse de vision tardive est possible. Les dépôts blanchâtres bilatéraux au niveau de la couche de Bowman sont situés dans l’aire de la fente palpébrale. En histologie, on observe des dépôts granuleux calciques (colorés par le rouge alizarine) et non calciques dans la couche de Bowman et le stroma antérieur (granules sphéroïdes en microscopie électronique à transmission).
Cette dystrophie correspond à une amylose cornéenne primitive (dépôts amyloïdes). La transmission est autosomique récessive. Le gène responsable, M1S1, est localisé en 1p32 [4]. La pathologie est fréquente au Japon.
Le début est précoce dans la petite enfance. La symptomatologie fonctionnelle associe une baisse d’acuité visuelle progressive, des érosions épithéliales récidivantes très douloureuses, une photophobie et un larmoiement.
Dans la forme typique en forme de mûre (mulberry type), l’examen met en évidence des dépôts nodulaires reflétant la lumière, translucides, centraux, en frai de grenouille, antérieurs (dépôts amyloïdes entre épithélium et couche de Bowman), ayant tendance à confluer avec le temps. Les autres formes cliniques sont des opacités en bande, des opacités stromales, la forme kumquat-like et les formes associées à une néovascularisation cornéenne périphérique.
En histologie, les dépôts amyloïdes sont situés entre l’épithélium et la couche de Bowman ainsi que dans le stroma antérieur.
Le traitement repose sur la kératoplastie lamellaire, mais la dystrophie récidive rapidement.
Elle est autosomique dominante ; le gène en cause est celui d’une protéine de la famille de la phosphoinositide kinase situé en 2q35 [4]. L’atteinte est bilatérale. La dystrophie est habituellement asymptomatique de découverte fortuite lors d’un examen ophtalmologique. L’examen montre des dépôts punctiformes blanchâtres ou annulaires diffus dans le stroma. En histologie, on observe la présence dans les kératocytes d’un matériel coloré par le bleu alcyan et le fer colloïdal (glycosaminoglycanes), mais également le noir Soudan (lipides complexes). En microscopie électronique à transmission, on note des vacuoles extracellulaires rondes, linéaires et curvilignes avec parfois un matériel dense, fibrillogranulaire, dans certains kératocytes et autour d’eux. Cette dystrophie ne requiert aucun traitement.
Elle est bilatérale et symétrique. La transmission est autosomique dominante à pénétrance complète. Elle se manifeste par un aspect de cornée laiteuse présent dès la naissance. L’absence d’atteinte endothéliale la distingue de la dystrophie endothéliale héréditaire congénitale. En histologie, on observe des lacunes stromales remplies de filaments et des dépôts colorés par le bleu alcyan. Cette dystrophie ne récidive pas après la greffe.
Elle est autosomique dominante, apparaît entre 30 et 60 ans [23]. Il s’agit d’un saupoudrage blanchâtre très fin et diffus, en avant de la membrane de Descemet. On observe : en OCT, une hyperréflectivité fine et fragmentée du stroma postérieur ; en microscopie confocale, de petites particules hyperréflectives dans le cytoplasme des kératocytes du stroma postérieur [1, 24]. Ces particules sont également retrouvées dans la matrice extracellulaire autour des kératocytes.
En histologie, on note des dépôts PAS positifs intracytoplasmiques dans les fibroblastes du stroma postérieur ; une membrane de Descemet normale.
La transmission est autosomique récessive. Le gène atteint (CYP4V2) est situé sur le chromosome 4q35. Il code pour une protéine de la superfamille des cytochromes P450 [4].
Le tableau clinique est avant tout marqué par une rétinopathie pigmentaire avec des microcristaux dans toutes les couches de la rétine. L’atteinte cornéenne est asymptomatique : dépôts cristallins jaunâtres dans le stroma antérieur de la cornée périphérique, sous la couche de Bowman.
En histologie, des cristaux biréfringents de 10 à 20 µm de diamètre sont mis en évidence dans le stroma cornéen, les kératocytes, la rétine, les fibroblastes conjonctivaux et cutanés et les lymphocytes circulants.
Citons la dystrophie mucineuse sous-épithéliale, la dystrophie discoïde centrale, la dystrophie de Pillat et la dystrophie centrale nuageuse de François.
• Collyres mouillants et cicatrisants. Ils sont utilisés dans la plupart de ces dystrophies quel que soit le stade évolutif.
• Photokératectomie thérapeutique au laser Excimer. Elle permet de traiter des opacités superficielles ne dépassant pas 150 µm. Les complications possibles sont un retard de cicatrisation, des infections, la récurrence de la dystrophie, le haze, l’hypermétropisation et l’astigmatisme irrégulier.
• Kératoplastie lamellaire antérieure automatisée ou profonde. C’est la technique de choix pour traiter la plupart des dystrophies stromales entraînant une baisse de vision importante pour lesquelles les possibilités de la photokératectomie sont dépassées. Elle n’est pas indiquée dans la dystrophie maculaire, car l’endothélium est atteint.
• Kératoplastie transfixiante.
Déficit en α-lécithine cholestérol acyltransférase de transmission autosomique récessive. Elle se manifeste cliniquement par une opacification cornéenne bilatérale et progressive donnant un aspect d’yeux de poisson bouilli, une anémie et une atteinte rénale. En histologie, on observe : des vacuoles optiquement vides dans le stroma et la couche de Bowman ; des dépôts colorés par le noir Soudan ; une diminution de la densité kératocytaire.
Anomalies génétiques à transmission autosomique récessive, elles se manifestent cliniquement par des dépôts cristallins dans le stroma antérieur sous forme d’aiguilles polychromatiques (fig. 9-33), associés à une atteinte conjonctivale, irienne, choroïdienne et rétinienne et une atteinte rénale. En histologie, on observe des cristaux de cystine intracellulaires dans toute l’épaisseur de la cornée.
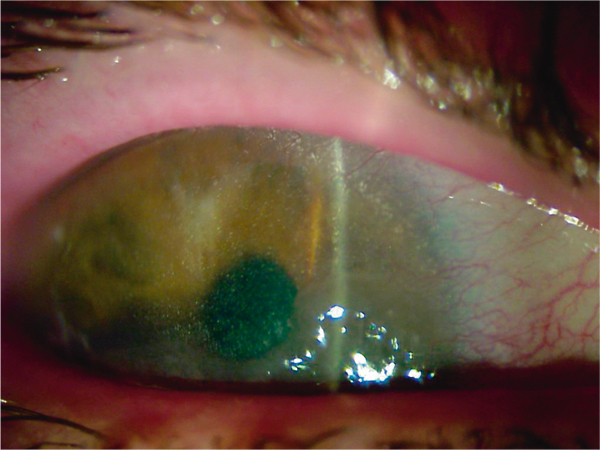
Fig. 9-33 Cystinose, aspect biomicroscopique.
Mutations du gène de la tyrosine aminotransférase à transmission autosomique récessive. Elle se manifeste cliniquement par des opacités cornéennes épithéliales et sous-épithéliales, des ulcérations, des érosions cornéennes récidivantes, une hypertrophie de la conjonctive tarsale, une hyperkératose palmoplantaire et une augmentation du taux de tyrosine dans le sang et les urines.
Déficits en enzymes de dégradation des mucopolysaccharides à transmission autosomique récessive sauf pour le syndrome de Hunter qui est récessif lié à l’X. Elles se manifestent cliniquement par une opacité cornéenne faite de petits points juxtaposés avec un retentissement relativement modéré sur la vision.
Syndrome de Hurler : mucopolysaccharidose I H, déficit en iduronidase, atteinte rétinienne et glaucome chronique, atrophie optique, nanisme, dysmorphie faciale, surdité, retard psychomoteur, hépatosplénomégalie, hernie ombilicale, insuffisance aortique, pronostic vital engagé précocement.
Syndrome de Scheie : mucopolysaccharidose I S, déficit en iduronidase, taille et espérance de vie normales, déformations des mains, surdité, insuffisance aortique, syndrome du canal carpien, hernie.
Maladie de Hurler-Scheie : mucopolysaccharidose I H/S, déficit en iduronidase, atteinte rétinienne et glaucome chronique, kystes arachnoïdiens, dysostose multiple, surdité, retard mental modéré, hernie, atteinte cardiaque, décès précoce.
Syndrome de Hunter : mucopolysaccharidose II, déficit en iduronidase sulfate sulfatase, atteinte rétinienne et des nerfs optiques, petite taille, hépatosplénomégalie, surdité, retard mental, atteinte cutanée, atteinte cardiaque, décès avant l’âge adulte.
Syndrome de Morquio : mucopolysaccharidose IV A, déficit en galactosamine-6-sulfatase, nanisme, dysostose multiple.
Syndrome de Maroteau-Lamy : mucopolysaccharidose VI A B, déficit en N-acétylglucosamine-4-sulfatase, atteinte des nerfs optiques, dysostose multiple, sténose aortique, hernie.
Syndrome de Sly : mucopolysaccharidose VII, déficit en β-glucuronidase, atteinte rétinienne et des nerfs optiques, retard psychomoteur, dysostose multiple, hépatosplénomégalie, hernie.
Déficit en α-mannosidase et α-fucosidase à transmission autosomique récessive. Tableau clinique proche de celui du syndrome de Hurler avec un déficit de l’immunité humorale et cellulaire et une cornea verticillata.
Déficit en acide homogentisique oxydase à transmission autosomique récessive. Dépôts cornéens superficiels pigmentés ressemblant à des gouttelettes d’huile noire, avec une atteinte cutanée et cardiaque.
Hyperlipoprotéinémie de type I (hyperchilomicronémie) : déficit en lipoprotéine lipase ou en apo C, autosomique dominant, kératopathie lipidique, xanthomes palpébraux, lipemia retinalis, hépatosplénomégalie, pancréatite.
Hyperlipoprotéinémie de type II (hyper-β-lipoprotéinémie) : autosomique dominante, kératopathie lipidique, arc cornéen, atteinte coronarienne.
Hyperlipoprotéinémie de type III : autosomique récessive, arc cornéen, lipemia retinalis, xanthélasmas palpébraux, xanthomes tubéreux, atteinte coronarienne, atteinte vasculaire périphérique, diabète, augmentation du cholestérol, des triglycérides et des β-lipoprotéines.
Hyperlipoprotéinémie de type IV (hyper-pré-β-lipoprotéinémie) : autosomique dominante, arc cornéen, lipemia retinalis, xanthélasmas palpébraux, xanthomes, atteinte coronarienne, atteinte vasculaire périphérique, diabète, goutte, augmentation du cholestérol, des triglycérides, des chilomicrons et des VLDL (very low density lipoprotein).
Mutations du gène ABC1. Elle associe opacification cornéenne, atteinte rétinienne, lagophtalmie avec kératite d’exposition, atteinte conjonctivale, neuropathie, hépatosplénomégalie, lymphadénopathie, hypertrophie amygdalienne.
[1] Borderie V. Imagerie de la cornée et de la surface oculaire. Paris : MedCom ; 2014.
[2] Rapuano CJ, Vajpayee RB. Corneal calcific band keratopathy. Curr Opin Ophthalmol 2011 ; 22 : 283-9.
[3] Cursiefen C, Colin J, Dana R, et al. Consensus statement on indications for anti-angiogenic therapy in the management of corneal diseases associated with neovascularisation : outcome of an expert roundtable. Br J Ophthalmol 2012 ; 96 : 3-9.
[4] Hurmeric V, Yoo SH, Karp CL, et al. In vivo morphologic characteristics of Salzmann nodular degeneration with ultra-high-resolution optical coherence tomography. Am J Ophthalmol 2011 ; 151 : 248-56.
[5] Roszkowska AM, Aragona P, Spinella R, et al. Morphologic and confocal investigation on Salzmann nodular degeneration of the cornea. Invest Ophthalmol Vis Sci 2011 ; 52 : 5910-9.
[6] Woodward M, Randleman JB, Larson PM. In vivo confocal microscopy of polymorphic amyloid degeneration and posterior crocodile shagreen. Cornea 2007 ; 26 : 98-101.
[7] Weiss JS, Khemichian AJ. Differential diagnosis of Schnyder corneal dystrophy. Dev Ophthalmol 2011 ; 48 : 67-96.
[8] Munier F, Schorderet D, Uffer S. Dystrophies héréditaires de la cornée. In : Dufier JL, Kaplan J. Œil et génétique. Paris : Masson ; 2005, p. 139-58.
[9] Paliwal P, Sharma A, Tandon R, et al. TGFBI mutation screening and genotype-phenotype correlation in north Indian patients with corneal dystrophies. Mol Vis 2010 ; 16 : 1429-38.
[10] Labbe A, Nicola RD, Dupas B, et al. Epithelial basement membrane dystrophy : evaluation with the HRT II Rostock Cornea Module. Ophthalmology 2006 ; 113 : 1301-8.
[11] Ogasawara M, Matsumoto Y, Hayashi T, et al. KRT12 mutations and in vivo confocal microscopy in two Japanese families with Meesmann corneal dystrophy. Am J Ophthalmol 2014 ; 157 : 93-102.
[12] Patel DV, Grupcheva CN, McGhee CN. Imaging the microstructural abnormalities of Meesmann corneal dystrophy by in vivo confocal microscopy. Cornea 2005 ; 24 : 669-73.
[13] Charukamnoetkanok P, Tuli S, Azar DT. Anterior corneal dystrophies : dystrophies of the epithelium, epithelial basement membrane, and Bowman’s layer. In : Smolin and Thoft’s the cornea : scientific foundations and clinical practice. 4th ed. Philadelphia : Lippincott Williams & Wilkins ; 2005, p. 813-24.
[14] Mastropasqua L, Nubile M, Lanzini M, et al. Corneal and conjunctival manifestations in Fabry disease : in vivo confocal microscopy study. Am J Ophthalmol 2006 ; 141 : 709-18.
[15] Wasielica-Poslednik J, Pfeiffer N, Reinke J, et al. Confocal laser-scanning microscopy allows differentiation between Fabry disease and amiodarone-induced keratopathy. Graefes Arch Clin Exp Ophthalmol 2011 ; 249 : 1689-96.
[16] Kobayashi A, Sugiyama K. In vivo laser confocal microscopy findings for Bowman’s layer dystrophies (Thiel-Behnke and Reis-Bucklers corneal dystrophies). Ophthalmology 2007 ; 114 : 69-75.
[17] Kuchle M, Green WR, Volcker HE, Barraquer J. Reevaluation of corneal dystrophies of Bowman’s layer and the anterior stroma (Reis-Bucklers and Thiel-Behnke types) : a light and electron microscopic study of eight corneas and a review of the literature. Cornea 1995 ; 14 : 333-54.
[18] Schneider S, Sorbara L, Jones L. Confocal microscopy and optical coherence tomography imaging of hereditary granular dystrophy. Cont Lens Anterior Eye 2010 ; 33 : 33-40.
[19] Rosenberg ME, Tervo TM, Gallar J, et al. Corneal morphology and sensitivity in lattice dystrophy type II (familial amyloidosis, Finnish type). Invest Ophthalmol Vis Sci 2001 ; 42 : 634-41.
[20] Fujiki K, Fujimaki T, Murakami A, Sugiyama K. In vivo laser confocal microscopic findings of corneal stromal dystrophies. Arch Ophthalmol 2007 ; 125 : 1168-73.
[21] Kawashima M, Kawakita T, Den S, et al. Comparison of deep lamellar keratoplasty and penetrating keratoplasty for lattice and macular corneal dystrophies. Am J Ophthalmol 2006 ; 142 : 304-9.
[22] Vesaluoma MH, Linna TU, Sankila EM, et al. In vivo confocal microscopy of a family with Schnyder crystalline corneal dystrophy. Ophthalmology 1999 ; 106 : 944-51.
[23] Klintworth GK. The molecular genetics of the corneal dystrophies-current status. Front Biosci 2003 ; 8 : 687-713.
[24] Kobayashi A, Ohkubo S, Tagawa S, et al. In vivo confocal microscopy in the patients with cornea farinata. Cornea 2003 ; 22 : 578-81.