Anomalies iriennes
K. Angioi, C.Orssaud
Aniridie et colobomes sont des conséquences d’un développement anormal de l’œil : anomalie de migration dans l’aniridie et anomalie de fermeture de la fente fœtale dans les colobomes. Ces anomalies de l’iris peuvent être isolées ou faire partie intégrante d’une dysgénésie du segment antérieur ou d’un syndrome.
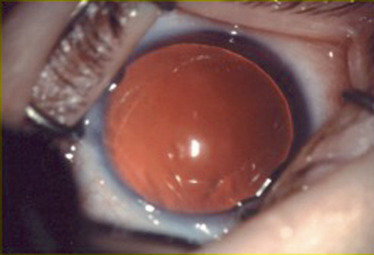
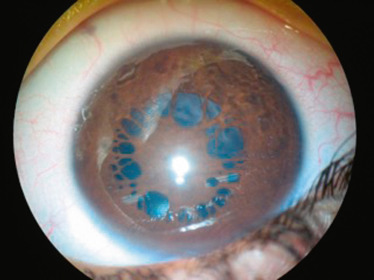
L’aniridie (Fig. 10-1) est une anomalie de développement de l’iris qui correspond à une malformation congénitale bilatérale, définie par l’absence totale ou quasi totale de l’iris. Elle peut être liée à une hérédité autosomique dominante (deux tiers des cas) avec une expression variable dans la même famille ou survenir de façon sporadique. Son incidence est évaluée entre 1/50 000 et 1/100 000. Les formes familiales ont une transmission autosomique dominante à pénétrance incomplète et expressivité variable. Il existe une mutation du gène PAX6 situé sur le bras court du chromosome 11. L’importance de ce gène au cours du développement explique que, même si l’aniridie est le signe d’appel le plus évident, il peut y avoir d’autres anomalies morphologiques associées qu’il faut savoir rechercher attentivement et systématiquement dans ce contexte. Les mutations du gène PAX6 ont en effet été identifiées dans un certain nombre d’anomalies du développement oculaire (cornée, cristallin, pupilles ectopiques, atteintes du segment antérieur, hypoplasie fovéale familiale isolée, atteinte du nerf optique).
Les cas sporadiques (sans antécédent familial connu) peuvent être isolés ou être intégrés dans un syndrome WAGR (Wilms tumor, Aniridia, Genital anomalies, mental Retardation) associant néphroblastome (tumeur de Wilms), aniridie, malformations génito-urinaires et retard mental. L’anomalie est située sur le chromosome 11(11p13). La délétion atteint le gène PAX6 (qui induit l’aniridie) et les gènes contigus dont celui du néphroblastome (WT1). Un caryotype doit être réalisé chez les enfants atteints d’une aniridie sporadique, car si la délétion est présente, le dépistage du néphroblastome doit être envisagé systématiquement et régulièrement du fait du risque important (25 à 30 % ).
Lors de l’examen, l’iris est absent dans sa quasi-intégralité. Il persiste souvent un petit résidu tissulaire en périphérie. L’équateur du cristallin est visible à travers l’iris ainsi que la zonule. Certaines formes plus frustes ont été décrites avec parfois seulement une disparition de la partie centrale de l’iris. L’atteinte irienne qui est le point d’appel majeur est souvent associée à des atteintes d’autres structures oculaires, avec en particulier l’atteinte de l’épithélium cornéen (dystrophie, opacités, ulcérations, néovascularisation) liée à une insuffisance limbique. Le cristallin peut être atteint sous forme d’une ectopie cristallinienne ou d’une cataracte polaire qui survient dans 50 à 80 % des cas avant l’âge de 20 ans. Le glaucome très fréquent (jusqu’à 75 % des patients avec l’âge) est secondaire à l’existence d’adhérences iriennes au niveau du trabéculum [1]. Il existe une hypoplasie fovéolaire associée qui est responsable d’une acuité visuelle souvent basse, voire très basse et d’un nystagmus. Une photophobie est souvent présente et peut être très gênante pour les patients.
Le traitement propose dans tous les cas une correction optique adaptée à la réfraction et la prescription de verres solaires pour limiter la photophobie. En cas de cataracte totale ne permettant plus l’accès au fond d’œil, une chirurgie est indiquée, mais la question de l’implantation éventuelle se discute. Le traitement du glaucome est particulièrement difficile du fait des anomalies structurelles (voir chapitre 12).
Le mot « colobome » définit un manque. Les colobomes peuvent concerner l’iris seul ou plusieurs autres structures comme le corps ciliaire, le cristallin ou la choriorétine. Ils sont liés à une anomalie de fermeture de la fente embryonnaire lors de l’embryogenèse (avec implication de mutations du gène PAX2). La localisation est de ce fait toujours dans le territoire inféronasal.
Au niveau de l’iris, il peut s’agir d’une atteinte partielle avec hypoplasie du feuillet antérieur de l’iris. Celui-ci apparaît alors aminci, atrophique ou hypopigmenté dans le quadrant nasal inférieur. Quand il est complet, la surface manquante est d’importance variable. Il se traduit par un défect irien plus ou moins étendu en nasal inférieur avec un aspect de déformation pupillaire classiquement dit en « trou de serrure » (Fig. 10-2a-c). Les petites atteintes peuvent être difficiles à mettre en évidence sur un iris foncé. Quand le colobome est assez large, l’équateur du cristallin est visible dans la zone du colobome. L’examen doit rechercher des anomalies associées :
une encoche cristallinienne : au niveau du cristallin l’aspect est celui d’un défect tissulaire mais il ne s’agit pas d’un colobome au sens propre du terme (différence embryologique); du fait de l’absence d’iris et souvent de corps ciliaire, il existe un relâchement de la zonule dans le secteur concerné aboutissant à un aspect de colobome cristallinien;
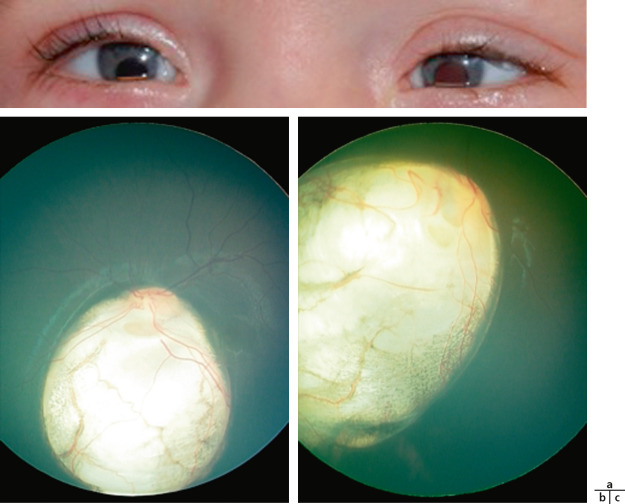
une atteinte choriorétinienne : le colobome est en fait initialement un colobome rétinien et c’est l’absence de développement rétinien qui aboutit à l’absence de développement de la choroïde. Il apparaît à l’examen du fond d’œil sous forme d’un territoire blanc jaunâtre, de taille variable dans le territoire inféronasal qui peut aller de la périphérie à la papille. Certains sont localisés à un petit territoire rétinien et d’autres peuvent atteindre le nerf optique (Fig. 10-3a-c).
Selon la taille du colobome irien, la symptomatologie va être plus ou moins marquée allant d’asymptomatique à une photophobie handicapante et une gêne esthétique pour les colobomes de grande taille. Le colobome irien peut être isolé ou associé à : des anomalies oculaires sévères comme une microphtalmie [2], un colobome papillaire ou papillochoroïdien; d’autres malformations organiques qu’il faut rechercher par un bilan complet surtout dans les formes bilatérales. Ainsi, le colobome irien peut :
entrer dans un cadre polymalformatif héréditaire; les plus fréquents sont les syndromes CHARGE (Coloboma, Heart defect, Atresia choanae, Retarded growth, Genital anomalies, Ear anomalies), de Rubinstein-Taybi, de Goldenhar, de Goltz, de Lenz, de Warburg, d’Aicardi;
être également lié à des anomalies chromosomiques : trisomies 13, 18 et 8; triploïdie; syndromes de l’œil de chat, de Turner, de Klinefelter.
Sur le plan fonctionnel, l’acuité visuelle dépend des anomalies oculaires associées (colobome uvéal, papillochoroïdien englobant ou non la macula). Le colobome irien isolé entraîne un astigmatisme généralement compris entre 2 et 4 D qui doit être corrigé pour ne pas entraîner une amblyopie fonctionnelle réfractive. Le colobome papillochoroïdien englobant la macula entraîne une amblyopie organique sévère le plus souvent associée à un strabisme et/ou un nystagmus sensoriel.
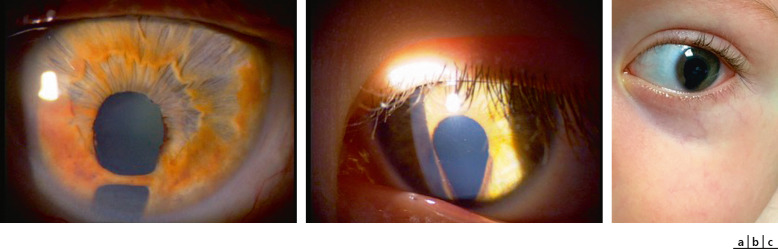
Fig. 10-2 a-c. Colobomes iriens unilatéraux.
La transillumination irienne est secondaire à l’altération de l’épithélium pigmenté. Les causes comprennent : albinisme, hypoplasie de l’iris, mégalocornée, syndrome de Marfan, cristallin et pupille ectopiques, microcorie, petit déficit irien transilluminable près de la racine de l’iris chez les yeux bleus sans retentissement clinique. Seul l’albinisme est abordé ici, car il s’agit de l’anomalie la plus importante.
On désigne sous le terme d’albinisme les atteintes héréditaires directement liées aux modifications de synthèse de la mélanine (pigment élaboré dans les mélanocytes). Il survient dans environ 1/20 000 naissances. L’albinisme est lié à un déficit qualitatif et quantitatif de mélanine par atteinte de la tyrosinase. La tyrosinase est l’enzyme qui permet la transformation de la tyrosine en mélanine. La mélanine est responsable de la pigmentation cutanée et des phanères et, au niveau oculaire, de la pigmentation de l’iris et de l’épithélium pigmentaire de la rétine. Les signes oculaires fréquents sont : une acuité visuelle basse avec un retard de maturation visuelle; un nystagmus; une hypoplasie fovéale; une hypopigmentation de l’iris responsable de la transillumination irienne; une photophobie; un strabisme; une stéréo-acuité réduite; une amétropie (astigmatisme, myopie ou hypermétropie); une anomalie des voies visuelles intracérébrales [3]. On distingue plusieurs types d’albinisme : la forme oculaire pure, la forme oculocutanée et les formes complexes qui s’accompagnent de pathologies mettant en jeu le pronostic vital (anomalie de la coagulation, immunodéficience, etc.).
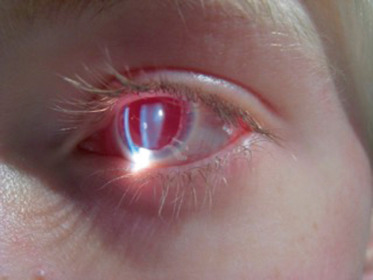
La forme oculaire, liée au gène OA1 est de transmission récessive liée à l’X. Le plus souvent, il existe un nystagmus lié à une mauvaise acuité visuelle et une photophobie plus ou moins marquée. L’iris est très clair, bleu clair ou gris clair. Lors de l’examen, on constate le phénomène de transillumination, c’est-à-dire la visibilité de la lueur orangée du fond d’œil à travers l’iris. Dans les formes peu marquées, il est plus facile de voir ce phénomène en regardant à côté du faisceau lumineux de la fente, et l’aspect est variable en rayons de roue ou ponctué (Fig. 10-4). Du fait de la très faible pigmentation, il est parfois possible de voir la zonule ou l’équateur du cristallin à travers l’iris. Le fond d’œil est également peu pigmenté, il n’y a pas de pigment maculaire et on constate la présence d’une hypoplasie fovéolaire. De ce fait, l’acuité visuelle est le plus souvent basse et entraîne la survenue d’un nystagmus. L’examen du fond d’œil est en général assez caractéristique mais la tomographie par cohérence optique (optical coherence tomography [OCT]) confirme l’hypoplasie fovéolaire. Si on réalise des examens électrophysiologiques dans le cadre du nystagmus, l’électrorétinogramme (ERG) et les potentiels évoqués visuels (PEV) peuvent être normaux, mais le plus souvent les PEV ont un aspect caractéristique pathognomonique : asymétrie des réponses croisées. Une partie des fibres temporales normalement directes décussent; de ce fait, en PEV, on obtient une diminution de la réponse corticale homolatérale à la stimulation, aboutissant à l’asymétrie des tracés.
Les formes oculocutanées liées à l’atteinte du gène AOC sont autosomiques récessives. Les signes ophtalmologiques sont les mêmes, auxquels viennent s’ajouter des signes cutanés avec une hypopigmentation de la peau et des phanères. La couleur de la peau est toutefois variable en fonction de l’ethnie du patient et il faut savoir penser au diagnostic chez des patients mélanodermes qui sont parfois moins pigmentés que leur famille.
Les formes complexes de l’albinisme peuvent être associées à des atteintes systémiques : les principaux diagnostics associés sont le syndrome de Chediak-Higashi (déficit immunitaire avec infections à répétition) et le syndrome de Hermansky-Pudlak.
Le traitement dépend de la sévérité de l’atteinte de l’affection et consiste à corriger au mieux la réfraction et à équiper les enfants d’une protection solaire en raison de la photophobie.
Tous les nouveau-nés naissent avec des iris peu pigmentés, donnant une teinte bleue. La couleur de l’iris définitive apparaît progressivement dans les mois suivant la naissance parallèlement à la multiplication plus ou moins importante des cellules pigmentées de l’iris : les cellules de Klumpenzellen et les chromatophores. Celles-ci sont localisées dans le stroma irien. Plus la quantité de ces cellules pigmentées est importante, plus l’iris a une teinte foncée. Toutes les altérations de la multiplication ou de la répartition de ces cellules peuvent entraîner une anomalie congénitale de coloration irienne. Les atteintes stromales secondaires sont quant à elles responsables de pertes plus ou moins étendues de ces cellules pigmentées à l’origine d’une anomalie acquise de la coloration de l’iris. Enfin, d’autres cellules pigmentées, au premier rang desquelles il faut citer les mélanocytes, peuvent venir modifier la couleur naturelle de l’iris en donnant des taches ou des plages plus foncées; c’est ce qui s’observe en présence de certains nævi iriens.
Il s’agit d’iris présentant des zones de coloration très contrastée, quelle que soit la taille de ces dernières. Cet aspect doit être différencié d’une variante de la normale de la coloration irienne qui n’est pas toujours homogène. Il faut également différencier cet aspect des anomalies iriennes observées au cours des dysgénésies du segment antérieur. La présence de défects localisés du tissu irien peut donner l’impression de territoires plus sombres lorsque l’examen est difficile chez un jeune enfant.
Mais une telle anomalie de la coloration irienne n’est généralement pas visible dès la naissance puisque la pigmentation irienne n’est pas encore développée. Elle apparaît au cours des premiers mois suivant la naissance dans les formes d’origine congénitale. Cette double coloration est retardée dans les formes acquises après une période au cours de laquelle l’iris avait un aspect normal.
Il existe plusieurs causes d’anomalie congénitale de la coloration irienne.
La description de ces taches varie beaucoup selon les auteurs.
Elles sont parfois décrites comme des plages d’iris normal au sein de zones d’atrophie irienne [4]. D’autres descriptions font état d’une couronne de points blancs ou jaunâtres de petite taille localisée à la périphérie de l’iris et plus classiquement au niveau de la jonction du tiers moyen et du tiers externe de l’iris. Quelle que soit la définition retenue, ces anomalies iriennes donnent une coloration irrégulière de l’iris. Ces lésions sont considérées comme étant pathognomoniques de la trisomie 21. Elles apparaissent tôt au cours de la vie et ont été rapportées chez de très jeunes enfants. Leur nombre semble ne pas varier avec l’âge.
Néanmoins, leur fréquence semble varier selon les populations. Ainsi, la présence de ces taches de Brushfield est rarement rapportée lors des études menées dans les populations asiatiques ou africaines [5]. Leur fréquence semble ne pas dépasser 5 % [4]. À l’inverse, ces lésions seraient beaucoup plus fréquentes dans les populations caucasiennes avec une fréquence variable selon les auteurs, généralement comprise entre 30 et 50 % , mais pouvant atteindre 90 à 100 % dans certaines séries. Cette différence de fréquence pourrait s’expliquer par la coloration de l’iris au sein de ces différentes populations. Ces taches de Brushfield doivent être recherchées à la lampe à fente et seraient d’autant plus faciles à observer que l’iris est peu coloré. En revanche, leur détection serait rendue difficile lorsque l’iris est foncé [5]. Mais, cette théorie est actuellement remise en question puisque des taches de Brushfield ont été retrouvées avec une fréquence identique à celle de l’Europe dans des pays où les iris sont volontiers foncés. Ces différences reposeraient sur des fonds génétiques variables selon les populations.
Une étude récente apporte une piste en mettant en évidence une corrélation entre la présence de taches de Brushfield et l’existence de troubles cardiaques congénitaux chez des patients porteurs de trisomie 21. Cette corrélation nécessite d’être confirmée par d’autres études.
Cet aspect particulier de l’iris, qui peut lui conférer un aspect bicolore, n’est pas pathognomonique du syndrome de Williams (ou syndrome de Williams-Beuren) mais il lui est volontiers associé [6]. Ce syndrome autosome dominant, décrit pour la première fois en 1961, est dÛ à une mutation génétique dans la bande 7q11.23. Ce syndrome est responsable de troubles cardiovasculaires avec anomalies des valves cardiaques et fragilité coronarienne rendant les enfants fragiles vis-à-vis de l’anesthésie qui doit être particulièrement précautionneuse. Il existe une déficience mentale modérée avec des troubles de l’attention. Néanmoins, ces enfants ont un grand sens musical. Ils présentent par ailleurs une dysmorphie. Un strabisme convergent est retrouvé chez 30 à 80 % des patients. Celui-ci est précoce dans près de la moitié des cas. Une amblyopie est fréquemment observée. Enfin, il existe une tortuosité vasculaire rétinienne anormale.
L’iris stellaire a longtemps été considéré comme étant la conséquence d’une hypoplasie du stroma irien [6]. En fait, chez les enfants porteurs du syndrome de Williams-Beuren, la portion antérieure de l’iris est anormale. La pigmentation de la portion pupillaire jusqu’à la collerette est normale comme la collerette elle-même. En revanche, cette collerette irienne peut être absente ou présenter une malposition, étant déplacée en périphérie. Cette localisation anormale lui donne un aspect sinueux. Plus en dehors, le stroma irien est parcouru de travées très particulières car plus radiaires que la normale, localisées à la surface du stroma irien. En extrême périphérie, l’iris antérieur est normal. Cet aspect particulier de l’iris est d’autant plus facile à observer que celui-ci est de coloration claire.
Plusieurs mécanismes sont responsables du changement de coloration de l’iris avec aspect bicolore au cours de l’enfance. Il faut citer tout d’abord certaines pathologies génétiques non tumorales (aspect bicolore dû à l’apparition de très nombreuses lésions colorées), mais aussi les tumeurs iriennes, bénignes ou non. Nous ne ferons que citer les inflammations uvéales des cyclites de Fuchs, du syndrome de Posner-Schlossman ou de l’uvéite herpétique qui peuvent être responsables d’atrophie irienne et donc de plages de dépigmentation.
Le xanthogranulome juvénile (XGJ) entre dans le cadre des histiocytoses non langerhansiennes, une classe de maladies histiocytaires [7]. Il se manifeste par la survenue de tumeurs fibro-histiocytaires bénignes parfois appelées nævo-xantho-endothéliomes. L’étiologie de cette pathologie rare est encore imprécise. Sa prévalence est inconnue. Elle toucherait plus de 50 % des enfants de moins de 3 ans avec une légère prédominance masculine. Le XGJ est généralement isolé mais s’observe avec une fréquence accrue chez les enfants porteurs de neurofibromatose de type 1 (NF1) [7].
La majorité des manifestations du XGJ est localisée au niveau de la peau et des muqueuses, avec des localisations extracutanées au niveau hépatique, cardiaque, splénique, rénal, au niveau du système nerveux central et surtout de l’œil.
Des tumeurs cutanées ou sous-cutanées apparaissent par vagues au cours des deux ou trois premières années de vie et parfois jusqu’à l’adolescence [8]. Elles sont bien délimitées arrondies ou ovalaires, de couleur jaunâtre prenant un aspect de papules fermes, élastiques, de quelques millimètres à 2 ou 3 cm de diamètre et d’épaisseur variable et pouvant saigner. Leur évolution spontanée est favorable sans traitement. Du fait du risque fonctionnel visuel du XGJ, un examen ophtalmologique systématique peut être recommandé.
L’atteinte oculaire intéresse principalement l’uvée. Elle est retrouvée dans environ 0,5 % des cas et serait d’autant plus fréquente que l’enfant est plus jeune et présente de nombreuses lésions cutanées [8]. Son pic de fréquence survient entre 1 et 2 ans [9]. Samara estime que les atteintes cutanée et oculaire sont rarement concomitantes [9]. Le XGJ se manifeste par une masse nodulaire brun jaunâtre, orangée ou transparente siégeant habituellement dans la portion moyenne de l’iris. Cette lésion peut être diffuse, donnant un aspect de fine lésion recouvrant les cryptes iriennes comme une toile constituée de mailles avec des élevures irrégulières (Fig. 10-5a, b). Une hétérochromie irienne ou une néovascularisation en regard du XGJ sont parfois retrouvées. Les complications sont volontiers révélatrices d’une localisation oculaire passée inaperçue [10]. La survenue d’une hyperhémie oculaire ou d’un hyphéma non traumatique est caractéristique et doit faire rechercher ce diagnostic chez le très jeune enfant. Cet hyphéma est plus ou moins important, parfois limité à du sang recouvrant la masse irienne, volontiers récidivant et peut s’associer à une hémorragie intravitréenne. Il existe un risque d’hypertonie oculaire, parfois très sévère pouvant altérer la fonction visuelle et imposant un traitement hypotonisant [11]. D’autres complications sont rapportées : infiltrats choroïdiens ou atrophie optique.
Le XGJ est également responsable de lésions limbiques ou conjonctivales dans moins de 10 % des cas. Celles-ci seraient l’apanage des adolescents et des adultes et réalisent des tumeurs régulières, jaunâtres, généralement uniques, qui ont tendance à progresser sur la cornée. Enfin, il a été rapporté des lésions palpébrales.
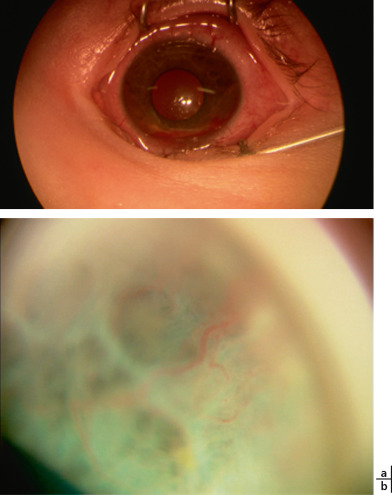
Fig. 10-5 a, b. Xanthogranulome juvénile irien avec hyphéma (grossissement en b).
(Remerciements au Pr D. Denis.)
Lorsque le XGJ de l’iris n’est pas visible, l’OCT du segment antérieur, l’ultrasound biomicroscopy (UBM) ou l’angiographie à la fluorescéine permettent de mettre en évidence la lésion irienne sous forme d’une fine pellicule épi-irienne [9].
Le diagnostic de certitude repose sur l’examen histologique après biopsie à l’aiguille d’une lésion intra-oculaire ou de l’exérèse d’une lésion cornéolimbique ou palpébrale. Il retrouve un infiltrat constitué de cellules histiocytaires spumeuses mononucléées, de lymphocytes, de cellules neutrophiles et macrophagiques et comportant les classiques cellules géantes de Touton : cellules polynuclées possédant un cytoplasme éosinophile au centre. Cet examen histologique permet le diagnostic différentiel et d’éliminer des masses uvéales ou cornéolimbiques associées à d’autres pathologies histiocytaires, lymphoprolifératives ou métastatiques.
Certains recommandent un traitement systématique des lésions iriennes pour éviter les complications (hyphéma, blocage de l’angle) [9]. Une corticothérapie topique ou péri-oculaire permettrait d’obtenir en quelques semaines une disparition de la lésion dans la majorité des cas. Un traitement hypotonisant oculaire doit être associé à la corticothérapie, avec une surveillance du nerf optique. Plus récemment, une équipe a utilisé avec succès l’injection d’anti-vascular endothelial growth factor (anti-VEGF) [12].
Les lésions cornéolimbiques et palpébrales nécessitent une exérèse chirurgicale. La prise en charge des lésions choroïdiennes ou orbitaires reste mal codifiée et peut reposer sur la radiothérapie, les corticoïdes ou l’exérèse.
Encore appelés corps de Lisch, ces hamartomes iriens s’observent au cours des NF1, détaillées dans le chapitre 26. Ils ne sont pas présents à la naissance et apparaissent au cours de la première décennie puisqu’ils sont retrouvés chez 42 % des enfants de moins de 4 ans et chez 55 % dans la tranche d’âge 5-6 ans [13]. Leur nombre peut être extrêmement variable, généralement inférieur à 15 et est indépendant de l’importance des autres signes cliniques de la NF1 [14]. Ils se présentent sous la forme de petites masses arrondies, posées sur l’iris, de coloration jaune à brun chamois [13]. Ces nodules de Lisch sont plus volontiers retrouvés dans la portion inférieure de l’iris et sont plus nombreux lorsque l’iris est clair [14]. Ils n’ont aucun retentissement sur la fonction visuelle.
La nature exacte de ces nodules reste mal connue. Certains auteurs évoquent la présence de cellules mélanocytaires, d’autres celle de cellules d’aspect proche des fibroblastes et ces lésions auraient une origine proche des schwannomes. Le rôle des rayons ultraviolets (UV) dans leur survenue reste controversé. Mais il expliquerait leur distribution prédominant en inférieur et leur apparition retardée. L’implication de modificateurs génétiques dans leur expression est probable et pourrait être liée soit à la mutation du gène responsable de la NF1, soit aux gènes responsables de la coloration irienne [14].
Lorsque la NF1 n’est pas connue, la présence de ces lésions caractéristiques doit en faire rechercher d’autres arguments cliniques et paracliniques. Leur présence est en effet considérée comme rare chez des sujets non atteints de NF1. La présence d’au moins deux nodules de Lisch constitue l’un des critères requis pour porter ce diagnostic lorsque celui-ci est simplement suspecté devant un aspect compatible avec des « taches café au lait » , ces dernières étant parfois peu spécifiques à cet âge [15].
Jackobiec, dans une étude portant sur 189 lésions pigmentées de l’iris et du corps ciliaire, constate qu’il n’existe pas de différence clinique permettant de différencier un nævus bénin (Fig. 10-6) d’un mélanome [16]. Seul leur potentiel évolutif permet de les différencier cliniquement. Un nævus peut augmenter de taille chez l’enfant, mais cette croissance est lente. L’échographie montre également l’absence de masse en cas de nævus. Mais il y a peu de données concernant l’apport de cette technique, y compris de l’UBM, pour différencier ces deux types de lésions.
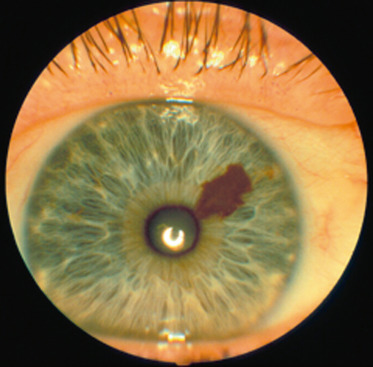
Les tumeurs malignes sont dominées par les mélanomes.
Les mélanomes de l’uvée de l’enfant représentent environ 1 à 2 % de l’ensemble des tumeurs mélaniques. Dans 25 à 60 % des cas, il s’agit de mélanomes iriens, lesquels sont exceptionnels avant l’âge de 6 ou 7 ans.
Un mélanome réalise une masse généralement pigmentée (Fig. 10-7) – les tumeurs achromes n’étant retrouvées que dans 20 % des cas – et nourrie par des vaisseaux anormaux. Il bombe dans la chambre antérieure dans la moitié des cas ou peut prendre un aspect en plaque voire, chez l’enfant, en tapioca. Le risque de métastase s’accroît avec l’augmentation d’épaisseur de cette masse. Ce mélanome est plus volontiers localisé dans le quadrant inférieur de l’iris et s’étend sur 2 à 4 secteurs. L’angiographie à la fluorescéine retrouve des vaisseaux anormaux. Dans un quart des cas, cette tumeur peut entraîner un glaucome ou, du fait des rétractions iriennes dont elle est responsable, des correctopies ou des ectropions de l’uvée. Il existe également un risque d’envahissement de l’angle iridocornéen, mais aussi de la sclère et du corps ciliaire, qui rend le pronostic péjoratif comme la présence de métastases à distance. L’UBM peut étudier l’épaisseur de cette tumeur et son envahissement.
Lorsque l’envahissement local est trop important, l’énucléation reste d’actualité. Mais des traitements conservateurs peuvent être proposés lorsque le mélanome est de petite taille : radiothérapie par plaque, protonthérapie ou iridectomie sectorielle réalisant une tumorectomie totale [17].
Nous ne ferons que mentionner les mélanomes du corps ciliaire qui ne représentent qu’un quart des mélanomes de l’uvée de l’enfant. Longtemps asymptomatiques, ils peuvent être évoqués devant une dilatation des veines épisclérales en regard de la tumeur, une hypotonie oculaire ou, à l’inverse, un glaucome. Ces mélanomes peuvent être découverts devant des modifications de la réfraction secondaire à une subluxation du cristallin qui au maximum peut s’opacifier. Le pronostic est souvent mauvais, avec une survie de 24 mois à 10 ans, en fonction du volume de la tumeur lors du diagnostic qui est corrélé au risque de métastase [18].
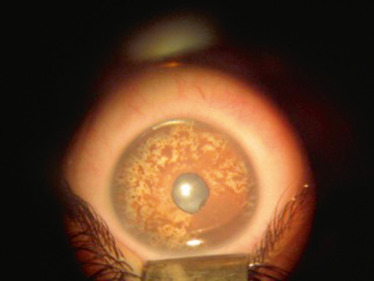
Encore appelée dyctiome, cette tumeur rare est retrouvée chez des enfants de moins de 9 ans [19]. Elle réalise une masse peu pigmentée, de couleur grise ou chamois, localisée au niveau du corps ciliaire ou de l’iris. Sa croissance est généralement assez lente, ce qui permet de la différencier d’un mélanome achrome. En échographie, elle apparaît comme une masse pleine, parfois comportant des kystes intratumoraux. Son évolution se fait vers une subluxation du cristallin qui peut se cataracter, mais aussi vers un glaucome « néovasculaire » du fait de la survenue de néovaisseaux iriens. Elle peut également évoluer vers un envahissement locorégional.
Histologiquement, cette tumeur est sans doute d’abord bénigne mais évoluerait vers une malignité plus ou moins importante [20]; elle est peu radio-sensible et récidive fréquemment après exérèse simple, ce qui explique que l’énucléation reste le traitement recommandé [19].
Les métastases iriennes chez l’enfant sont exceptionnelles. Les rares cas rapportés concernent des lymphomes qui infiltrent l’iris en entraînant une hétérochromie ou un nodule. Le diagnostic doit être évoqué devant une pseudo-uvéite qui ne cède pas sous un traitement habituel.
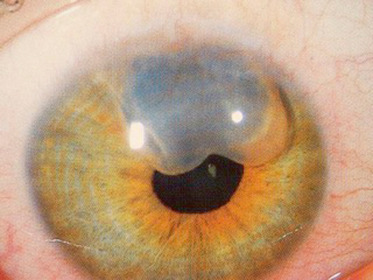
L’iris peut être le siège de kystes longtemps asymptomatiques. Lorsqu’ils grossissent, ils entraînent une voussure localisée de l’iris (Fig. 10-8) et peuvent fermer l’angle iridocornéen. Mais, quand ils sont proches du rebord pupillaire, ils peuvent venir dans l’axe pupillaire. Quand ils ne sont pas visibles lors de l’examen clinique, l’échographie UBM permet de préciser leur structure et l’échographie en mode B permet de vérifier qu’il ne s’agit pas de l’extension d’une tumeur. Le plus souvent congénitaux, certains kystes sont acquis ou traumatiques et alors plus volontiers évolutifs. Leur nature et leur constitution diffèrent en fonction de leur localisation.
Leur origine reste discutée. Il s’agirait d’une évagination du feuillet postérieur de l’épithélium pigmenté irien ou d’un clivage entre les deux feuillets de cet épithélium du fait de la persistance d’un espace embryonnaire. Hormis quand ils sont périphériques, ces kystes sont très pigmentés en transillumination et possèdent une paroi très échogène entourant un centre liquidien en UBM. Ils sont généralement bilatéraux et multiples quand ils ont une localisation intermédiaire.
Appelés « disloqués » , les kystes provenant du sulcus peuvent migrer dans le vitré ou en chambre antérieure.
Translucides, les kystes du stroma sont congénitaux ou pigmentés s’ils sont acquis. L’UBM retrouve un kyste liquidien dont la paroi est fine s’ils ne sont pas pigmentés (Fig. 10-9).
Ces kystes sont translucides, localisés dans le stroma et leur diagnostic est porté par l’échographie qui retrouve une paroi à plusieurs couches entourant un centre hyperéchogène.
Tant qu’ils sont asymptomatiques, ces kystes ne nécessitent aucun traitement, car ils ont tendance à involuer avec le temps. Lorsqu’ils entraînent une hypertonie par blocage de l’angle iridocornéen, il faut intervenir en excisant le kyste. Le risque est la récidive.
Deux formes d’hétérochromies coexistent : une différence de couleur entre l’iris de chaque œil ou l’iris bicolore. Il est difficile de différencier les deux formes dans la littérature. À côté de l’exceptionnelle hétérochromie congénitale autosomique dominante et des pathologies que nous avons évoquées plus haut, d’autres affections peuvent entraîner des modifications unilatérales de l’iris. Citons les syndromes de Claude-Bernard-Horner congénitaux ou apparus avant l’âge de 2 ans qui sont responsables d’une hypochromie unilatérale du côté atteint (voir plus loin). Les syndromes de Waardenburg de type II et IV peuvent également être responsables d’une hétérochromie moins par inhomogénéité de la couleur de chaque iris que par différence de coloration entre chaque œil [21].
Cette anomalie constitue l’un des aspects du syndrome de persistance de la vascularisation fœtale décrit par Goldberg. Elle correspond à la non-résorption de la tunica vasculosa lentis anterior (TVLA). Celle-ci se développe au cours de la 6e semaine de vie embryonnaire alors que se met en place le cristallin. Cette tunique vasculaire commence à disparaître dès le 6e mois de la vie embryonnaire grâce à la mise en jeu de mécanismes apoptotiques et a totalement disparu au cours du 8e mois [22, 23]. Lorsque les mécanismes apoptotiques sont défaillants, il peut persister un reliquat membranaire plus ou moins important appelé « membrane pupillaire » . Celle-ci est facilement visible; elle est uni- ou bilatérale et d’importance variable. Son étude histologique a permis de montrer qu’elle est composée de tissu irien normal, y compris vasculaire, avec parfois un taux élevé de fibrocytes et de fibres de collagène réalisant une membrane fibrovasculaire et pouvant expliquer la résistance à l’apoptose [24].
Cette membrane pupillaire serait retrouvée chez plus de 90 % des nouveau-nés.
Dans la grande majorité des cas, la membrane pupillaire est très discrète, limitée à la présence de quelques fibres iriennes tendues d’une portion de la collerette irienne à une autre. Ces fibres laissent l’axe visuel dégagé ou, si elles le traversent, n’entraînent aucun retentissement visuel. Une régression spontanée, parfois totale, peut s’observer au cours de la première année de vie chez un nouveau-né à terme [23].
Plus rarement, la membrane pupillaire est plus dense et importante, elle est retrouvée chez 4 à 5 % des nouveau-nés arrivés à terme [25]. Cette fréquence est plus élevée chez les enfants prématurés puisque la régression de la TVLA n’est pas encore terminée. Elle va se poursuivre à un rythme normal dans les semaines suivant la naissance prématurée [22]. Dans la majorité des cas, cette membrane n’entraîne aucun retentissement visuel. En effet, elle n’est pas homogène mais constituée d’extensions tubulaires tendues de la collerette irienne vers la pupille (Fig. 10-10). Ces extensions tubulaires se rejoignent au niveau d’une plaque plus ou moins grande, souvent décentrée par rapport à l’axe optique et percée de plusieurs opercules [25]. Certaines de ces extensions tubulaires gagnent la cornée ou la cristalloïde antérieure à laquelle la plaque peut adhérer sur une surface plus ou moins importante. Les adhérences cornéennes présentent un risque d’œdème cornéen ou d’opacification cornéenne localisée, alors que les adhérences cristalliniennes peuvent entraîner des cataractes [24]. Seules les formes sévères requièrent une prise en charge thérapeutique chirurgicale qui doit être exceptionnelle. Dans tous les cas, il y a un risque d’amblyopie organique et fonctionnelle anisométropique par astigmatisme induit. Cela justifie un traitement d’amblyopie par occlusion controlatérale et instillation homolatérale d’un collyre mydriatique permettant de dégager l’axe visuel et, éventuellement, de faire céder certaines insertions sur la collerette irienne ou lâcher les adhérences cristalliniennes [22, 25]. Ce traitement doit être accompagné d’une prise en charge des anomalies réfractives et de l’amblyopie associées.
Dans d’autres cas, la membrane pupillaire est dense, recouvrant la totalité de l’axe pupillaire [22]. Le risque d’amblyopie et de strabisme est majeur. Différentes approches ont été proposées pour dégager l’axe visuel :
réalisation d’une néopupille dans la membrane à l’aide d’un laser argon ou d’un laser YAG (yttrium aluminium garnet) [26]; les complications de cette technique sont la cataracte, l’inflammation et l’hypertonie oculaire [24, 26]; ce traitement simple peut être indiqué chez les grands enfants mais le problème est qu’à un âge avancé, l’amblyopie est installée;
ablation chirurgicale de la membrane constituant la technique de choix chez les enfants plus jeunes : après un abord limbique, l’injection de la substance viscoélastique permet le plus souvent de décoller la membrane pupillaire de la cristalloïde antérieure, puis de la découper à l’aide de microciseaux; si un fragment reste adhérent en dehors de l’axe visuel, certains auteurs conseillent de le laisser lorsqu’il existe un risque de léser le cristallin [24].
Les risques spécifiques de cette intervention chirurgicale sont la survenue d’une cataracte et d’un hyphéma lorsque la membrane pupillaire est vascularisée. L’hyphéma peut également survenir au décours de la pupilloplastie au laser YAG. Néanmoins, quelques cas rapportés font état d’hyphémas spontanés à partir d’un reliquat vasculaire n’ayant pas régressé.
Les membranes pupillaires sont soit isolées (généralement partielles) ou associées (généralement obturantes) à d’autres malformations oculaires (microcornée, cornea plana, cataracte, colobome rétinien ou kératocône postérieur, etc.) ou générales (tétralogie de Fallot, etc.) rapportées dans la littérature [26]. Leur prise en charge dépend de l’atteinte associée.
Bien que le terme de « myosis congénital » soit parfois utilisé, il semble préférable de parler de « microcorie congénitale » pour définir les petites pupilles d’un diamètre inférieur à 2 mm dues à une anomalie de développement du muscle dilatateur de l’iris et transmises selon un mode autosomique dominant [27]. Du fait du caractère bilatéral de cette atteinte, il n’y a pas d’anisocorie. Aucune dilatation, ou une très faible dilatation, est obtenue après installation de collyre mydriatique. La surface irienne est anormalement plate, peu colorée et l’iris est irrégulièrement transilluminable. Il s’y associe une dysgénésie de l’angle iridocornéen, responsable du glaucome juvénile observé, et des anomalies réfractives à type de myopie et d’astigmatisme [27].
La microcorie congénitale est parfois isolée, mais il a été rapporté des microcories avec d’autres dysgénésies du segment antérieur de l’œil, tel un syndrome de Rieger. Enfin, cette anomalie s’observe lors du syndrome de Pierson qui associe atrophies irienne et du corps ciliaire, anomalies cristalliniennes, malvoyance, hypotonie et syndrome néphrotique.
L’anisocorie passe volontiers inaperçue chez les enfants dont l’iris est foncé. Néanmoins, l’utilisation de photoscreeners dans le dépistage de troubles réfractifs facilite leur mise en évidence puisque certains de ces appareils donnent le diamètre pupillaire [28].
Le défi, face à une anisocorie, consiste à déterminer si un bilan est nécessaire ou si une simple surveillance suffit. Malheureusement, il n’existe pas de réel consensus récent ni de données d’evidence-based medicine dans la littérature concernant les tests à utiliser pour analyser cette anomalie du jeu pupillaire ou la conduite à tenir face à elle [29-32].
Lors de la découverte d’une anisocorie, il faut d’abord éliminer une anomalie pupillaire congénitale ou acquise qui modifie le jeu pupillaire : un colobome irien, une correctopie, des synéchies iridocristalliniennes ou iridocornéennes (secondaires à une dysgénésie du segment antérieur ou post-uvéitique). Une aniridie, notamment si elle n’est pas totale, peut être confondue avec une mydriase congénitale. Un examen attentif, éventuellement sous anesthésie générale en cas de doute, permet alors de rétablir le diagnostic.
Lorsque le diagnostic d’anisocorie est posé, il convient de déterminer la pupille pathologique et, par conséquent, le contingent du système nerveux autonome potentiellement altéré. L’analyse des variations du diamètre pupillaire et de l’anisocorie à la lumière et à l’obscurité permet théoriquement de répondre à cette question. C’est la pupille dont le diamètre change le moins lors des variations d’éclairement qui est pathologique. En présence d’une mydriase, l’anisocorie augmente à la lumière puisque la pupille pathologique ne se referme pas au fort éclairement. En revanche, en présence d’un myosis, l’anisocorie augmente à l’obscurité puisque la pupille pathologique ne se dilate pas ou mal dans cette ambiance lumineuse réduite. Rappelons qu’en présence d’une anisocorie physiologique, la différence de diamètre pupillaire reste inchangée quelles que soient les conditions d’éclairement. Cette analyse reste difficile lorsque les iris sont foncés ou chez le très jeune enfant dont le jeu pupillaire peut être lent. Nous reviendrons sur l’intérêt des tests pharmacologiques.
Il faut, parallèlement à cette analyse, demander aux parents les conditions et la date de découverte de cette anisocorie, le déroulement de la grossesse et de l’accouchement ainsi que les antécédents de l’enfant, notamment traumatiques. Il est important de noter un éventuel changement de comportement de l’enfant ou toute anomalie neurologique associée. Chez les grands enfants, une éventuelle instillation de collyre ou de toxiques est recherchée.
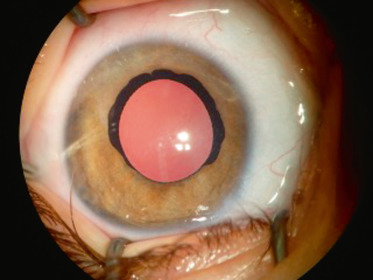
L’anisocorie congénitale doit être découverte ou présente sur des photographies avant 5 mois pour certains auteurs ou au cours de la première année de vie pour d’autres [30, 31]. Mais ce critère n’est pas formel puisqu’une anisocorie « acquise » peut survenir avant l’âge d’un an expliquant que la définition reste floue.
La mydriase congénitale est exceptionnelle et s’observe au cours de quelques syndromes rares par mutation du gène ACTA2, par anomalies congénitales cardiaques ou dysplasie septo-optique. En dehors de ces situations, il faut systématiquement évoquer une cause acquise (voir plus loin) y compris lorsque cette mydriase apparaît très tôt au cours de la vie.
Un myosis peut exister isolément ou entrer dans le cadre d’un syndrome de Claude-Bernard-Horner (CBH) congénital qui représente la première cause d’anisocorie de l’enfant [31]. La définition clinique de celui-ci varie dans la littérature, rendant difficile la détermination d’un consensus concernant la prise en charge d’un myosis congénital.
Ce syndrome est dÛ à une atteinte de l’un des trois neurones de la voie du sympathique oculaire entre l’hypothalamus et l’œil [32]. Le diagnostic du CBH est facile en présence d’un myosis responsable d’une anisocorie qui s’aggrave à l’obscurité et d’un ptosis très peu important n’atteignant pas l’axe visuel [32]. L’existence d’une hypochromie irienne du côté atteint (témoignant d’un CBH apparu avant l’âge de 2 ans) et d’une anhydrose de l’hémiface homolatérale complète volontiers le tableau dans les formes congénitales. Certains auteurs considèrent qu’une hétérochromie est nécessaire pour parler de CBH congénital. La réduction de la fente palpébrale responsable d’un aspect d’enophtalmie par élévation de la paupière inférieure est rarement retenue dans la littérature. Lorsque tous ces symptômes sont retrouvés le diagnostic de CBH est certain.
L’existence d’un traumatisme obstétrical constitue la cause principale du CBH congénital [30]. Ce diagnostic, retrouvé à l’interrogatoire et renforcé par une éventuelle atteinte du plexus brachial, nécessite une simple surveillance sachant qu’une pathologie sous-jacente peut se révéler. Les autres causes de CBH congénital sont représentées par les malformations carotidiennes congénitales, le neuroblastome, l’astrocytome du tronc cérébral et les adénopathies cervicales d’origine inflammatoire. Une élévation franche du taux des catécholamines urinaires est un argument en faveur d’un neuroblastome. Cet examen est maintenant réalisable sur un échantillon d’urine et ne nécessite plus le recueil sur 24 heures [33]. Mais un taux normal ne permet pas d’éliminer formellement ce diagnostic [30, 33]. Face à un CBH congénital isolé, sans anomalie générale lors de l’examen clinique (avec palpation des aisselles, du cou et de l’abdomen à la recherche d’une éventuelle tumeur), plusieurs équipes ont comparé la pratique d’une neuro-imagerie systématique (imagerie par résonance magnétique [IRM]; scanner de la tête, du cou et du thorax) à une simple surveillance [30-32]. Ces études retrouvent une cause tumorale chez moins de 10 % des enfants, les autres cas correspondant à des CBH congénitaux idiopathiques. C’est pourquoi, bien que le risque de survenue d’un neuroblastome ne soit pas nul, Smith recommande une simple surveillance clinique après un dosage des catécholamines urinaires en reconnaissant la limite de cet examen [30]. Néanmoins, dans tous les cas, un bilan neuroradiologique s’impose s’il apparaît une hétérochromie ou une aggravation d’une anhydrose (témoignant d’une atteinte du 3e neurone) [32]. Pour d’autres auteurs, la réalisation précoce d’une IRM reste systématiquement nécessaire [34]. Des neuroradiologues ont proposé de réaliser une échographie cervicale à la recherche d’un neuroblastome.
Lorsqu’il n’existe qu’un myosis congénital isolé, se pose la question de l’intérêt des épreuves pharmacologiques pour confirmer ou éliminer l’existence d’un CBH. Le test à la cocaïne (2,5 à 10 % ) est souvent indispensable à ce diagnostic dans la littérature anglo-saxonne. De fait, certains auteurs considèrent comme un « CBH » l’association « myosis et test à la cocaïne positif » [31, 32, 34]. Mais il est parfois difficile de disposer de ce collyre en dehors d’un milieu hospitalier. Le test à l’apraclonidine à 0,5 % peut constituer une bonne alternative, du moins chez un enfant de plus de 1 an, puisque ce collyre est contre-indiqué en usage thérapeutique chez le jeune enfant [35].
Il n’existe aucune donnée dans la littérature concernant la conduite à tenir devant un myosis congénital strictement isolé. Il semble logique de considérer qu’il n’existe aucun risque d’atteinte du sympathique oculaire si le myosis est isolé et que les épreuves pharmacologiques sont négatives. Un suivi annuel est alors suffisant. En revanche, si les épreuves pharmacologiques n’ont pas été réalisées ou si elles sont positives, il faut considérer qu’il existe une « suspicion de CBH congénital » en l’absence d’autres signes de la triade clinique (myosis, ptosis, diminution de la fente palpébrale). Dans un tel cas et en l’absence d’argument clinique en faveur d’une lésion tumorale ou d’une adénopathie, il est possible de surseoir à un bilan neuroradiologique sous réserve d’une surveillance rapprochée. Au moindre doute, celui-ci devra être demandé.
Nous ne ferons que citer les anisocories post-traumatiques qui nécessitent un bilan ophtalmologique et neuroradiologique systématique.
Une mydriase acquise doit dans un premier temps faire rechercher une atteinte de la 3e paire crânienne devant un ptosis ou une paralysie/parésie oculomotrice. Cette association doit faire pratiquer un bilan neuroradiologique en urgence, au mieux une IRM couplée à une angiographie à la recherche d’une tumeur cérébrale ou d’une malformation vasculaire : anévrisme, angiome, etc.
Lorsque la mydriase est isolée, il faut réaliser un test à la pilocarpine diluée à 0,125 % . La fermeture de la pupille pathologique à l’instillation de ce collyre signe une atteinte du ganglion ciliaire ou au-delà (on dit que « la mydriase s’inverse » ). La littérature anglo-saxonne parle de «adie tonic pupil » . Celle-ci peut s’accompagner de troubles de l’accommodation, d’une hyperréactivité pupillaire en vision de près et de mouvements vermiculaires de l’iris. Cette anomalie s’observe dans les suites de maladies infectieuses ou après des crises de migraine [36]. Il faut en rapprocher les mydriases cycliques récidivantes, considérées comme des équivalents migraineux chez l’enfant. Cette pupille tonique n’est pas systématiquement réversible et peut se compliquer d’amblyopie puisque l’œil atteint a perdu la capacité d’accommoder et de corriger son hypermétropie. Si le test à la pilocarpine diluée est négatif, la normalisation de la pupille pathologique après installation de pilocarpine à 1 % permet d’évoquer une atteinte parasympathique isolée qui nécessite également de réaliser un bilan neuroradiologique urgent. Celui-ci peut retrouver une fissuration d’un anévrisme supraclinoïdien dont le risque est faible chez l’enfant, de l’ordre de 7 % . Ce dernier survient volontiers dans un contexte post-traumatique ou au cours d’une dysplasie vasculaire.
Enfin, si l’instillation de pilocarpine est négative, il faut évoquer des causes iatrogènes : par exemple, pollen de datura ou patch de scopolamine. Mais, les enfants peuvent s’instiller tout collyre laissé à leur portée.
À cet âge, les étiologies sont principalement post-traumatiques ou post-chirurgicales, infectieuses ou tumorales (neuroblastome, rhabdomyosarcome, tumeur du tronc cérébral) [30, 31]. Les causes vasculaires semblent rares dans la littérature en pédiatrie. Bien que les étiologies des CBH acquis et congénitaux ne soient pas totalement identiques, une même prise en charge peut être évoquée.
L’anisocorie physiologique est fréquente. Elle est définie comme la présence d’une différence d’au moins 0,4 mm entre les pupilles des deux yeux, mais rarement supérieur à 1,3 mm, valeur qui doit faire rechercher un autre mécanisme [37], même s’il est admis que 18 à 20 % des patients présentent une telle différence de taille de pupille. De plus, une cause physiologique est retenue pour environ 15 % des cas d’anisocorie tous âges confondus. Une fréquence identique a été retrouvée lors d’examens réalisés avec un photoscreener chez plus de 18 % des enfants [28]. Normalement, l’anisocorie physiologique varie peu lors du passage de la lumière à l’obscurité. Néanmoins, il peut exister des fluctuations du diamètre pupillaire au cours de la journée. De plus, cette anisocorie physiologique se majore parfois à l’obscurité pouvant faire suspecter un CBH. Les tests pharmacologiques permettent théoriquement de rétablir le diagnostic, car ils sont négatifs en cas d’anisocorie physiologique.
Les déformations d’une pupille entière peuvent réaliser une correctopie (déplacement le plus souvent bilatéral et symétrique de la pupille) ou une dyscorie (déformation pupillaire en fente pouvant évoquer une pupille de chat). Certaines de ces déformations pupillaires sont parfois génétiquement déterminées et peuvent entrer dans le cadre de syndromes rares, tel le syndrome « ptosis-strabisme-correctopie » observé en fait dans une seule famille, etc. Elles sont alors congénitales, parfois mais non obligatoirement bilatérales. Il faut rappeler que le cat-eye syndrome s’accompagne d’un colobome pupillaire et non pas d’une dyscorie.
Ces déformations pupillaires peuvent également être la conséquence d’une traction ou d’une déformation irienne, parfois congénitale, qu’il faut s’appliquer à mettre en évidence.
Le dictionnaire de l’Académie de médecine rappelle qu’au cours d’une polycorie vraie, chaque orifice pupillaire doit être entouré d’un contingent musculaire et se contracter à la lumière. Cette situation est exceptionnelle et les mécanismes de formation de ces pupilles multiples restent peu clairs [38]. Il a été évoqué la fermeture précoce d’un colobome avec du tissu ecto- et mésodermique, une anomalie lors de la formation du cristallin durant la vie embryonnaire, aboutissant à la constitution de ponts iriens, ou à l’individualisation d’une déformation irienne durant l’embryogenèse.
Cette anomalie associe une hyperplasie de l’épithélium pigmenté de l’iris qui déborde sur la face antérieure de celui-ci au niveau d’un secteur pupillaire plus ou moins étendu, une atrophie du stroma irien et, pour certains auteurs, une dysgénésie de l’angle iridocornéen avec une insertion antérieure de la racine de l’iris (Fig. 10-11). C’est la raison pour laquelle cette anomalie s’accompagne volontiers d’un glaucome congénital ou juvénile. Elle s’observe également au cours de deux principaux syndromes : le syndrome de Prader-Willi et la NF1 [39].
[1] Nezzar H, Chiambaretta F, Rigal D, et al. L’iris et sa pathologie. Rapport annuel des Sociétés de Paris. Novembre Marseille: Lamy (2003). 65-80
[2] Nischal KK Pediatric iris anomaly Pediatric ophthalmology and strabismus : Oxford University Press (2015).
[3] Lee AG, Geloloneck ML, Pau DC Pupil anomalies and reactions Pediatric Ophthalmology and Strabismus Saunders: Elsevier (2013). 393-399chapitre 40.
[4] Afifi HH, Abdel AAA, El-Bassyouni HT, et al. Distinct ocular expression in infants and children with Down syndrome in Cairo, Egypt : myopia and heart disease JAMA Ophthalmology 2013 ; 131 : 1057-1066
[5] Liza-Sharmini AT, Azlan ZN, Zilfalil BA Ocular findings in Malaysian children with Down syndrome Singapore Medical Journal 2006 ; 47 : 14-19
[6] Holmstrom G, Almond G, Temple K, et al. The iris in Williams syndrome Archives of Disease in Childhood 1990 ; 65 : 987-989
[7] Haroche J, Abla O Uncommon histiocytic disorders : Rosai-Dorfman, juvenile xanthogranuloma, and Erdheim-Chester disease Hematology Am Soc Hematol Educ Program 2015 ; 2015 : 571-578
[8] Cypel TKS, Zuker RM Juvenile xanthogranuloma : case report and review of the literature Can J Plast Surg 2008 ; 16 : 175-177
[9] Samara WA, Khoo CTL, Say EA, et al. Juvenile xanthogranuloma involving the eye and ocular adnexa : tumor control, visual outcomes, and globe salvage in 30 patients Ophthalmology 2015 ; 122 : 2130-2138
[10] Niu L, Zhang C, Meng F, et al. Ocular juvenile xanthogranuloma Optom Vis Sci 2015 ; 92 : e126-e133
[11] Harvey P, Lee JA, Talbot JF, Goepel JR Isolated xanthogranuloma of the limbus in an adult Br J Ophthalmol 1994 ; 78 : 657-659.
[12] Ashkenazy N, Henry CR, Abbey AM, et al. Successful treatment of juvenile xanthogranuloma using bevacizumab J AAPOS 2014 ; 18 : 295-297
[13] Lubs ML, Bauer MS, Formas ME, Djokic B Lisch nodules in neurofibromatosis type 1 N Engl J Med 1991 ; 324 : 1264-1266
[14] Boley S, Sloan JL, Pemov A, Stewart DR A quantitative assessment of the burden and distribution of Lisch nodules in adults with neurofibromatosis type 1 Invest Ophthalmol Visual Sci 2009 ; 50 : 5035-5043
[15] Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference Archives of Neurology 1988 ; 45 : 575-578
[16] Jakobiec FA, Silbert G Are most iris « melanomas’really nevi? A clinicopathologic study of 189 lesions Archives of Ophthalmology (Chicago, Ill: 1960) 1981 ; 99 : 2117-2132
[17] Tran TA, Hirst LW, Axelsen RA, Welch R Iris melanoma in 12-year-old boy Clin Exp Ophthalmol 2006 ; 34 : 489-490
[18] Yousef YA, Alkilany M Characterization, treatment, and outcome of uveal melanoma in the first two years of life Hematolo Oncol Stem cell Ther 2015 ; 8 : 1-5
[19] Ramasubramanian A, Shields CL, Kytasty C, et al. Resection of intraocular tumors (partial lamellar sclerouvectomy) in the pediatric age group Ophthalmology 2012 ; 119 : 2507-2513
[20] Verdijk RM On the classification and grading of medulloepithelioma of the eye Ocul Oncol Pathol 2016 ; 2 : 190-193
[21] Winship I, Beighton P Phenotypic discriminants in the Waardenburg syndrome Clinical Genetics 1992 ; 41 : 181-188
[22] Burton BJ, Adams GG Persistent pupillary membranes Br J Ophthalmol 1998 ; 82 : 711-712
[23] Fard AM, Asghari S, Pourafkari L, Nader ND Persistent pupillary membrane QJM 2016 ; 109 : 139-140
[24] Iida M, Mimura T, Goto M, et al. Letter to the editor Excision of congenital bilateral persistent pupillary membrane in a child with exotropia Open ophthalmol J 2015 ; 9 : 33-35
[25] Doyle JJ, Reddy AK Bilateral persistent pupillary membranes JAMA Ophthalmology 2016 ; 134 : e160030
[26] Ahmad SS, Binson C, Lung CK, Ghani SA Bilateral persistent pupillary membranes associated with cataract Digit J Ophthalmol 2011 ; 17 : 62-65
[27] Rouillac C, Roche O, Marchant D, et al. Mapping of a congenital microcoria locus to 13q31-q32 Am J Hum Genet 1998 ; 62 : 1117-1122
[28] Silbert J, Matta N, Tian J, et al. Pupil size and anisocoria in children measured by the plusoptiX photoscreener J AAPOS 2013 ; 17 : 609-611
[29] Cahill JA, Ross J Eye on children : acute work-up for pediatric Horner’s syndrome. case presentation and review of the literature J Emerg Med 2015 ; 48 : 58-62
[30] Smith SJ, Diehl N, Leavitt JA, Mohney BG Incidence of pediatric Horner syndrome and the risk of neuroblastoma : a population-based study Archives of Ophthalmology (Chicago, Ill 1960) 2010 ; 128 : 324-329
[31] Jeffery AR, Ellis FJ, Repka MX, Buncic JR Pediatric Horner syndrome J AAPOS 1998 ; 2 : 159-167
[32] George ND, Gonzalez G, Hoyt CS Does Horner’s syndrome in infancy require investigation? Br J Ophthalmol 1998 ; 82 : 51-54
[33] Mahoney NR, Liu GT, Menacker SJ, et al. Pediatric horner syndrome : etiologies and roles of imaging and urine studies to detect neuroblastoma and other responsible mass lesions Am J Ophthalmol 2006 ; 142 : 651-659
[34] Liu GT, Mahoney NR, Avery RA, et al. Pediatric horner syndrome Archives of Ophthalmology (Chicago, Ill : 1960) 2011 ; 129 : 1108-1109author reply 1109.
[35] Chen PL, Hsiao CH, Chen JT, et al. Efficacy of apraclonidine 0.5 % in the diagnosis of Horner syndrome in pediatric patients under low or high illumination Am J Ophthalmol 2006 ; 142 : 469-474
[36] Sobreira I, Sousa C, Raposo A, et al. Ophthalmoplegic migraine with persistent dilated pupil J Child Neurol 2013 ; 28 : 275-276
[37] Suh SH, Suh DW, Benson C The Degree of anisocoria in pediatric patients with Horner syndrome when compared to children without disease J Pediatric Ophthalmol Strabismus 2016 ; 53 : 186-189
[38] Islam N, Mehta JS, Plant GT True polycoria or pseudo-polycoria? Acta Ophthalmol Scand 2007 ; 85 : 805-806
[39] Laaks D, Freeman N Congenital iris ectropion uveae presenting with glaucoma in infancy J AAPOS 2013 ; 17 : 214-216