Dialogue entre ophtalmologistes et pédiatres – interdisciplinarité
Coordonné par D. Denis, E. Bui Quoc, B. Chabrol , J. -M. Triglia
P. Wary, E. Bui Quoc, E. Zanin, M. Beylerian, J. -M. Triglia,D. Denis
L'œil est un organe complexe, et la diversité des tissus oculaires reflète en un seul organe celle de tout l'organisme. Il peut être le siège de malformations congénitales ou héréditaires, ou être atteint, par odre de fréquence, par des phénomènes infectieux, inflammatoires, tumoraux, traumatiques, toxiques ou paranéoplasiques :
- – les malformations oculaires congénitales sont dues à des perturbations des interactions cellulaires et des mécanismes moléculaires et peuvent concerner toutes les structures de l'œil. La morphogenèse oculaire commence à la 4 semaine de vie embryonnaire, lorsque l'ébauche oculaire s'individualise à partir des diverticules latéraux du cerveau antérieur; divers feuillets de l'embryon vont contribuer à l'élaboration du globe oculaire : le neurectoderme, l'ectoderme de surface, le mésoderme et les cellules de la crête neurale. Ainsi les dérégulations des processus embryologiques vont être à l'origine des malformations oculaires congénitales isolées ou syndromiques impliquant d'autres organes;
- – les maladies oculaires héréditaires sont dues à des mutations géniques identifiées par les techniques de séquençage à haut débit et peuvent être également isolées ou syndromiques impliquant aussi d'autres organes.
Toutes ces causes mettent en évidence la nécessité d'échanges cliniques indispensables entre l'ophtalmologie et d'autres spécialités pédiatriques, en d'autres termes d'une interdisciplinarité effective permanente dans notre pratique quotidienne.
Ainsi, la prise en charge d'une pathologie à la fois pédiatrique et ohptalmologique peut impliquer de nombreuses spécialités et l'ophtalmopédiatre va avoir un double rôle :
- – la recherche des signes ophtalmologiques pouvant orienter le diagnostic étiologique lors du diagnostic initial;
- – la mise en évidence d'une atteinte oculaire spécifique au cours du suivi, conséquence de la pathologie elle-même ou de sa thérapeutique.
Ces échanges interdisciplinaires vont avoir pour objectif d'obtenir, face à une atteinte oculaire et d'un autre organe, un diagnostic rapide avec mise en place d'un traitement adapté dans les meilleurs délais, afin de maintenir la fonction visuelle (dans ce contexte d'atteinte multiviscérale), tout en traitant la pathologie organique et/ou systémique associée.
L'interdisciplinarité en ophtalmologie pédiatrique est un sujet très vaste.
Les principaux contextes pathologiques abordés sont par exemple :
- – contexte d'urgences ou semi-urgences médicales en particulier infectieuses (herpès cornéen du nouveau-né, cellulites orbitaires, etc.), inflammatoires (rhumatismes et uvéites) et tumorales (rétinoblastome, rhabdomyosarcome, gliomes, etc.);
- – contexte neuropédiatrique, l'œil du fait de son origine embryologique commune avec le système nerveux (neuroépithélium) pouvant contribuer à l'expression clinique d'une pathologie neurologique ou neuro-ophtalmologique (hypertension intracrânienne, neurofibromatose, etc.);
- – contexte dermatologique, expliqué par l'origine embryologique neuro-ectodermique commune à la peau et aux structures oculaires; ainsi, une anomalie de différenciation cellulaire au cours de l'embryogenèse, par exemple du fait d'une expression anormale de médiateurs cellulaires comme des fibroblast growth factors (FGF) ou des transforming growth factors β (TGF-β), peut entraîner, selon les caractères de l'atteinte, des maladies affectant uniquement l'œil, la peau ou le cerveau, ou bien des maladies communes à ces différents organes (la neurofibromatose est un parfait exemple de ces maladies de la différenciation de l'ectoderme);
- – contexte de maladie générale ou systémique (oncologique, hématologique, endocrinologique, hépato-gastro-entérologique, cardiologique, pneumologique, néphrologique, etc.), de pathologie congénitale ou acquise avec recherche de complication oculaire secondaire à la maladie ou de complication iatrogène (par exemple, corticothérapie au long cours pouvant induire une cataracte et/ou une hypertonie oculaire; maladie du greffon contre l'hôte avec atteinte oculaire suite à une greffe de moelle osseuse, etc.);
- – contexte de pathologie locorégionale, l'orbite étant un carrefour entre crâne et face, concernant ainsi la pathologie oto-rhino-laryngo-logique (ORL), maxillofaciale ou neurochirurgicale; il s'agit parfois d'urgences chirurgicales, en cas par exemple de traumatisme craniofacial, d'hypertension intracrânienne ou de paralysie du VI aiguë;
- – contexte pédopsychiatrique en particulier dans les autismes et les troubles des apprentissages, etc.;
- – contexte anesthésique en préopératoire en vue d'une chirurgie chez un enfant fragile ou à risque (syndrome CHARGE, prématurés, syndromes polymalformatifs, etc.).
Les lignes directrices de ces dialogues établis entre ophtalmologistes et pédiatres spécialisés mettent l'accent sur :
- – les pathologies communes dont la prise en charge thérapeutique peut être urgente, semi-urgente ou différée et concerner les nouveau-nés, les nourrissons ou les enfants plus grands;
- – les thérapeutiques spécifiques de l'enfant, certaines pouvant nécessiter une hospitalisation pour des raisons d'observance ou de voie d'administration du médicament (forme injectable). Des protocoles de traitement et de surveillance sont établis sur fiche pratique pour certaines questions.
Ces dialogues abordent les thèmes suivants :
- – infectiologie et ophtalmologie;
- – rhumatologie et ophtalmologie;
- – neuropédiatrie et ophtalmologie;
- – dermatologie et ophtalmologie;
- – oncologie et ophtalmologie;
- – hématologie et ophtalmologie;
- – endocrinologie et ophtalmologie;
- – hépato-gastro-entérologie et ophtalmologie;
- – cardiologie et ophtalmologie;
- – pneumologie et ophtalmologie;
- – néphrologie et ophtalmologie;
- – ORL et ophtalmologie;
- – chirurgie maxillofaciale et ophtalmologie;
- – neurochirurgie et ophtalmologie;
- – pédopsychiatrie et ophtalmologie;
- – anesthésie pédiatrique – hypnose et ophtalmologie.
Ces dialogues montrent l'importance de connaître les interconnexions entre ces spécialités. Ils soulignent la nécessité d'une excellence dans la synergie requise entre les différentes disciplines pour une coordination la plus efficace possible entre pédiatres et ophtalmologistes.
En conclusion, face à une pathologie ophtalmologique et/ou pédiatrique, les objectifs de l'ophtalmologiste, en lien avec les pédiatres, doivent être la mise en place d'un dépistage, d'un diagnostic, d'un traitement adapté et d'un suivi sans délai, pour obtenir d'une part la parfaite prise en charge au long cours de la pathologie systémique, et pour garantir d'autre part la meilleure acuité visuelle possible pour une bonne qualité de vie et une bonne insertion sociétale.
E. Bosdure, M. Callet, E. Zanin, D. Denis
Toutes les structures de l'œil de l'enfant peuvent être atteintes par les agents infectieux. Cependant la majorité des infections pédiatriques est dominée par les infections virales et bactériennes, les infections fongiques et parasitaires étant relativement plus rares.
L'infectiologue a besoin de l'ophtalmologiste pour toute recherche de localisation ophtalmologique d'une pathologie infectieuse systémique ou locorégionale.
De nombreux micro-organismes peuvent être à l'origine d'infections transmissibles de la mère à l'enfant et responsables de pathologies fœtales graves tels que : Toxoplasma gondii, rubéole, cytomégalovirus etherpes simplex virus.
La toxoplasmose est dépistée de manière systématique et la séroconversion surveillée chez les femmes séronégatives afin de proposer un traitement rapide en cas de primo-infection destiné à limiter la transmission du virus au fœtus. La toxoplasmose congénitale concerne 1 enfant sur 10 000 naissances. En cas d'infection fœtale avérée, le traitement devra associer deux antiparasitaires (pyriméthamine et sulfadiazine). Le nouveau-né contaminé même asymptomatique devra être traité pendant 1 an. Le tableau clinique est variable en fonction de l'âge de contamination : avortement, mort fœtale in utero, calcifications intracrâniennes, micro- et macrocéphalies, hydrocéphalie, hépatosplénomégalie, cardiomégalie, ascite, retard de croissance, etc. L'atteinte ophtalmologique choriorétinienne est caractéristique et peut être isolée.
La séroprévalence de la rubéole est élevée en France, mais les infections maternofœtales sont rares (< 10 cas/an). En cas d'infection maternelle avant 12 semaines d'aménorrhée (SA), le risque malformatif est majeur et une interruption de grossesse peut être envisagée. Le syndrome de rubéole congénital associe : retard de croissance, atteinte oculaire (cataracte, choriorétinite, etc.), atteinte ORL, atteinte cardiaque. La fœtopathie associe un retard de croissance, des malformations multiples par embryofœtopathie (microcéphalie, microphtalmie, micrognathie, hypoplasie dentaire), et des atteintes polyviscérale et hématologique. La présence d'immunoglobulines M (IgM) chez le nouveau-né atteste d'une primo-infection. La prévention repose sur la vaccination des femmes en âge de procréer.
Le dépistage systématique du cytomégalovirus (CMV) durant la grossesse n'est pas recommandé en France. On estime à 50 % la proportion de femmes immunisées et à 0,5 % l'incidence de la primo-infection durant la grossesse soit 1500 cas/an. La réactivation du virus pendant la grossesse peut être un risque pour le fœtus. L'infection est généralement asymptomatique chez la mère. Chez le fœtus, elle peut entraîner : retard de croissance, hépatosplénomégalie, purpura, ictère, atteinte neurologique, choriorétinite. Pour les formes graves, la mortalité est de 20 à 30 % , et 90 % des survivants ont des séquelles graves neurologiques ou sensorielles. En cas de suspicion à la naissance d'infection à CMV, des prélèvements seront réalisés : la présence d'IgM dans le sang du nouveau-né confirme le diagnostic et un titre élevé est un reflet de la gravité de l'atteinte. En cas de forme sévère, un traitement par ganciclovir sera proposé pendant 21 jours.
La maladie est causée par un virus de la famille des Arenaviridae. En milieu extérieur, le virus survit dans les matières fécales de rongeurs. Il se transmet à l'homme par morsure ou contact entre les muqueuses ou une peau lésée et des déjections ou de la salive infectées. Une contamination par inhalation est possible [1, 2]. Chez l'adulte immunocompétent, l'infection peut passer inaperçue ou entraîner un syndrome pseudo-grippal; rarement elle peut se compliquer de méningo-encéphalite avec des signes neurologiques. En cas d'infection maternofœtale, différentes atteintes en fonction de l'âge de contamination peuvent se produire : avortement, fœotopathie avec choriorétinite, hydrocéphalie, macro- ou microcéphalie, retard mental, épilepsie. On note 35 % de mortalité chez les fœtus infectés et 60 % d'atteinte neurologique ou visuelle chez les survivants. La prévalence de cette infection est probablement sous-estimée chez les nourrissons atteints de retard mental et de baisse visuelle profonde. Les tests sérologiques sont généralement utilisés. Ils reposent sur la méthode enzyme-linked immunosorbent assay (ELISA) et peuvent être confirmés par Western blot. Leur positivité reste un argument diagnostique indirect. Nous ne disposons pas de traitement spécifique [1, 3]. Il faut noter que l'infection peut survenir chez les enfants plus âgés. Il s'agit alors de lésions principalement ophtalmologiques sans atteinte cérébrale significative. La négativité de la sérologie serait un argument pour le caractère acquis de l'infection.
Les kératoconjonctivites néonatales surviennent avant le 28 jour de vie et doivent faire craindre un germe issu d'infections sexuellement transmissibles ou IST (gonocoque et Chlamydia trachomatis) [4-6]. Nous observons actuellement une recrudescence des IST et notamment des infections gonococciques dans les pays industrialisés, y compris en France. Cette constatation a motivé la rédaction en 2010 de recommandations de l'Agence française de sécurité sanitaire des produits de santé (Afssaps) sur la prophylaxie des infections conjonctivales du nouveau-né. Rappelons que les infections gonococciques sont le plus souvent asymptomatiques chez la femme justifiant un prélèvement vaginal en cas de signe physique découvert à l'examen au spéculum, complété par un prélèvement systématique entre 35 et 38 SA. Ce dépistage permet de déterminer les nouveau-nés « à risque » qui devront bénéficier d'une prophylaxie. Ces risques sont clairement établis : parents à risque d'IST (antécédents, prélèvement en faveur) ou grossesse non ou mal suivie. La prophylaxie recommandée est : instillation de 1 goutte de rifamycine collyre dans les 2 yeux à la naissance.
La conjonctivite à Chlamydia touche 1,1 à 1,4 nouveau-né pour 1 000 naissances. Trente à 50 % des nourrissons dont la mère présente une infection génitale à Chlamydia non traitée peuvent présenter une conjonctivite et 10 à 20 % une pneumopathie. L'atteinte ophtalmologique survient entre le 3 et le 15 jour de vie : conjonctivite purulente avec fausses membranes, œdème palpébral, hypérémie majeure. Le diagnostic est confirmé par polymerase chain reaction (PCR) sur grattage conjonctival. Le traitement est urgent en raison des risques de cicatrices cornéennes et doit passer par voie systémique : érythromycine 50 mg/kg/j pendant 14 jours ± collyres à la quinolone et traitement de la mère.
La conjonctivite à Neisseria gonorrhoeae est plus rare (0,4 nouveau-né pour 1 000 naissances) mais plus grave. Elle apparaît dans les 5 premiers jours de vie : conjonctivite purulente très inflammatoire, membranes séro-hémorragiques bilatérales. Le diagnostic peut être confirmé par PCR sur grattage conjonctival mais ne doit pas retarder la mise en œuvre du traitement en extrême urgence devant le risque majeur d'opacification et de perforation cornéenne. Le traitement est systémique : céfotaxime (Claforan, 100 mg/kg/j) ou ceftriaxone (Rocéphine, 50 à 100 mg/kg/j) pendant 7 jours + antibiotique local et traitement de la mère.
Les infections virales sont dominées par le groupe des herpes viridae : herpès virus, virus zona-varicelle (VZV) et CMV. On trouve essentiellement deux types d'atteintes ophtalmologiques : les kératoconjonctivites et les uvéites. La localisation oculaire est particulièrement fréquente lors de la varicelle clinique et de l'herpès néonatal. L'herpès néonatal concerne 1 nouveau-né pour 7 500 naissances, touche davantage les jeunes primipares. Il s'agit du virus herpès simplex de type 2 (génital dans 70 % des cas) avec un contage fœtomaternel. La contamination se fait in utero par dissémination hématogène transplacentaire en cas de primo-infection maternelle avec virémie élevée ou par voie ascendante en cas de rupture prématurée des membranes. Il existe les risques suivants : avortement spontané, accouchement prématuré, retard de croissance in utero (RCIU), lésions cutanées in utero, microcéphalie, calcification intracérébrale, hydrocéphalie, hémorragie cérébrale, choriorétinite, microphtalmie, cataracte, aplasie maculaire ou papillaire, chorioamniotite. La contamination per-partum se fait lors du passage dans la filière génitale, le risque est maximal en cas de primo-infection récente (40 à 70 % d'atteinte). L'infection survient avant le 28 jour. La forme locale survient vers le 6 jour et associe une atteinte cutanée, buccale et oculaire. Les formes graves peuvent être localisées au système nerveux central avec une méningo-encéphalite ou disséminées avec une atteinte hépatique, pulmonaire et neurologique. Les signes ophtalmologiques typiques sont : vésicules herpétiques au bord palpébral; conjonctivite uni- ou bilatérale peu sécrétante non purulente, sérosanglante, compliquée d'une kératite ponctuée superficielle ou de microdendrites plutôt limbiques. Cette kératite peut se compliquer d'ulcère, d'opacification cornéenne, voire d'une choriorétinite [7]. Le traitement devra être systémique par aciclovir intraveineux, 250 à 500 mg/m toutes les 8 heures pendant 2 à 4 jours, accompagné d'un traitement local (aciclovir pommade 5 fois/j à adapter à l'examen clinique journalier) et d'un traitement de la mère.
Concernant le VZV chez l'enfant, la primo-infection sous forme de varicelle se complique rarement d'uvéite ou de vascularite rétinienne. Le zona ophtalmique de l'enfant est moins fréquent que chez l'adulte. Il peut survenir si la varicelle a eu lieu très tôt dans l'enfance et on considère ainsi que 5 à 10 % des zonas surviennent ainsi avant 25 ans [8]. En dehors des vésicules palpébrales, qui peuvent poser des problèmes d'irritation cornéenne mécanique ou de surinfection bactérienne, les manifestations oculaires sont le plus souvent de nature immunitaire et surviennent au décours de l'éruption cutanée. Des kératites stromales, des endothélites ou des uvéites sont donc possibles, les rétinites à VZV avec vascularites occlusives sont exceptionnelles. Il semblerait que l'hypoesthésie cornéenne séquellaire soit plus fréquente chez l'enfant. L'uvéite liée au virus VZV peut être observée en cas de varicelle ou de zona ophtalmique. Les présentations cliniques des uvéites à VZV et herpes simplex virus (HSV) sont proches : atteinte aiguë, unilatérale, plastique ou granulomateuse, synéchiante associée à une atrophie sectorielle de l'iris (plus importante pour HSV) et une hypertonie parfois majeure [1]. La vaccination anti-VZV est actuellement disponible. Très récemment, Naseri [9] a rapporté le cas d'un enfant de 9 ans, immunisé par le vaccin vivant atténué et développant une conjonctivite, une sclérite antérieure diffuse et une uvéite antérieure relativement sévère. Cet épisode est apparu 3 ans après la vaccination et la souche virale identifiée était le VZV sauvage et non la souche vaccinale, ce qui permet de relativiser l'efficacité d'une telle vaccination tout en éliminant, a priori, un rôle déterminant joué par le virus atténué (encadré 27-1).
Conduite à tenir devant un herpès néonatal
Herpès néonatal :
1 nouveau-né pour 7 500 naissances ;
herpes simplex virus de type 2 avec un contage foetomaternel ;
contamination per-partum lors du passage dans la filière génitale avec risque maximal en cas de primo-infection récente (40 à 70 % d’atteinte).
Apparition des signes cliniques d’infection le premier mois de vie :
forme locale avec atteinte cutanée, buccale et oculaire (autour du 6e jour) ;
formes graves, localisées au système nerveux central avec une méningo-encéphalite, ou disséminées avec atteinte hépatique, pulmonaire et neurologique.
Signes ophtalmologiques typiques :
vésicules herpétiques au bord palpébral ;
conjonctivite uni- ou bilatérale peu sécrétante non purulente, sérosanglante ;
kératite ponctuée superficielle ou microdendrites plutôt limbiques ;
ulcère cornéen pouvant se compliquer d’opacification cornéenne ;
choriorétinite.
Traitement de l’enfant en hospitalisation pédiatrique :
systémique : l’aciclovir per os (Zovirax® en solution buvable) a une absorption digestive limitée ; ainsi la forme intraveineuse sera choisie en cas d’atteinte sévère : aciclovir intraveineux 250 à 500 mg/m2 toutes les 8 heures en 3 perfusions pendant 2 à 4 jours (avec surveillance des effets indésirables, en particulier toxicité vasculaire de la perfusion), puis relais par valaciclovir (Zelitrex®) per os en préparation magistrale adaptée au poids de l’enfant (hors autorisation de mise sur le marché à cet âge) ;
local :
-
lavages,
aciclovir pommade 5 fois/j (Zovirax®) à adapter à l’examen clinique journalier (avec surveillance des effets indésirables),
traitement cicatrisant : pommade vitamine A.
Traitement de la mère.
N.B. : il n’existe aucune recommandation précise dans la littérature concernant un éventuel traitement préventif des récidives.
En cas d'infection disséminée bactérienne (par exemple septicémie sur endocardite), virale (par exemple infection à CMV) ou mycotique (par exemple candidémie), l'infectiologue fait appel à l'ophtalmologiste afin de rechercher une localisation oculaire. Cette dernière peut être choriorétinienne (par exemple syndrome des taches blanches diffuses, nodules de Roth dans le cadre d'une endocardite), vasculaire (vascularite occlusive), papillaire (œdème du nerf optique), vitréenne (endophtalmie endogène). L'ophtalmologiste pourra également être sollicité dans le cadre de la découverte d'une tuberculose à la recherche d'une uvéite granulomateuse, du classique tubercule de Bouchut ou de granulomes choroïdiens.
L'ophtalmologiste fait appel à l'infectiologue pour l'introduction et le suivi d'un traitement antiviral systémique. En effet, lorsque l'infection virale oculaire se complique et met en jeu le pronostic visuel, soit par la sévérité de l'atteinte soit par le problème des récurrences, un traitement par voie systémique est nécessaire. L'instauration du traitement est difficile selon l'âge de l'enfant du fait de la galénique non adaptée nécessitant l'aide de l'infectiologue pédiatre : aciclovir (Zovirax) intraveineux, valaciclovir (Zelitrex) per os à adapter au poids de l'enfant en préparation pharmaceutique. En raison des effets secondaires possibles des antiviraux au long cours, une surveillance biologique doit être organisée (fonction rénale, hépatique et numération tous les 3 à 6 mois) ainsi qu'une surveillance étroite de la toxicité locale des traitements topiques [10]. À noter qu'il semblerait que l'absorption digestive de l'aciclovir en sirop soit mauvaise, c'est pourquoi il faudra préférer la voie intraveineuse en cas d'atteinte sévère avec un relais par valaciclovir per os. Les discussions pourront également porter sur la dose préventive à mettre en place et la durée du traitement en cas de récurrences en l'absence de recommandations consensuelles.
Chez l'enfant plus grand, les kératites herpétiques ne sont pas exceptionnelles. Il s'agit le plus souvent d'une kératite stromale (dans plus de 60 % des cas, contre 20 % chez l'adulte) ou d'une kératite épithéliale dendritique avec une atteinte plus volontiers bilatérale (exceptionnelle chez l'adulte) et un risque de récurrence élevé : un enfant sur deux ferait une récurrence tous les 2 ans [7]. Ces atteintes peuvent entraîner une taie cornéenne, une néovascularisation cornéenne, un astigmatisme irrégulier et une amblyopie chez les enfants de moins de 6 ans.
Les uvéites liées aux Herpesviridae sont les premières causes d'uvéites aiguës virales. L'atteinte survient classiquement lors d'une réactivation virale et très rarement lors d'une primo-infection, le plus souvent liées à HSV-1. Il semble exister une corrélation entre l'âge et le type de virus. Ainsi, l'HSV-2 est particulièrement fréquent chez l'enfant, alors que l'HSV-1 est plus rencontré chez l'adulte jeune [1].
L'uvéite peut parfois être associée à une kératite ou à des vasculites rétiniennes en l'absence d'immunodépression [11]. L'examen ophtalmologique peut orienter le diagnostic sur le type de virus en cause (selon l'existence et le type de précipités rétrocornéens, le type de l'atrophie irienne, etc.). La confirmation du diagnostic peut être exigée dans les formes complexes par la réalisation d'une PCR sur un prélèvement d'humeur aqueuse par ponction de chambre antérieure, qui sera réalisée au bloc opératoire et sous anesthésie générale chez l'enfant.
Il s'agit d'une pathologie très fréquente, estimée selon les études à 14 cas pour 100 000 habitants. Chez l'enfant, la bactérie la plus fréquemment en cause est Haemophilus influenzae (45 à 60 % selon les séries) puis Streptococcus pneumoniae – pneumocoque (20 à 30 % ) avec des variations saisonnières : l'incidence des conjonctivites à pneumocoque est plus fréquente en hiver contrairement à Haemophilus influenzae plus fréquente au printemps et en été. La plupart du temps, les conjonctivites de l'enfant sont prises en charge par l'ophtalmologiste, cependant il peut faire appel à l'infectiologue lorsque la conjonctivite est associée à des signes systémiques. Les manifestations systémiques en cas de conjonctivite à Haemophilus influenzae peuvent être : malaise, fièvre, otite moyenne aiguë (syndrome conjonctivite-otite présent dans 25 à 30 % des cas), infection des voies respiratoires supérieures, épiglottite, pneumopathie, péricardite, arthrite, sinusite. Localement, la conjonctivite peut se compliquer de cellulite orbitaire, voire de méningite (souche encapsulée).
Les manifestations systémiques en cas de conjonctivite à Streptococcus pneumoniae sont essentiellement respiratoires : symptômes respiratoires bénins, rares pneumonies. L'infection à pneumocoque se fait à partir de la flore endogène ou par l'intermédiaire d'une contamination aérienne. La dissémination inter-humaine est favorisée par la promiscuité : crèche, école. Streptococcus pneumoniae est également responsable d'otite moyenne aiguë, de sinusite, d'angine érythémateuse, de bronchopneumopathie et c'est la deuxième cause de méningite chez l'enfant. Sur le plan ophtalmologique, la période d'incubation est d'environ 2 jours, la conjonctivite est maximale 2 à 3 jours après son début. Des hémorragies sous-conjonctivales ainsi qu'un chémosis sont fréquemment associés à l'atteinte conjonctivale.
À noter que la maladie des griffes du chat par infection à Bartonella henselae peut se présenter sous la forme d'une conjonctivite unilatérale en cas d'inoculation conjonctivale par pelage contaminé, avec lymphadénopathie préauriculaire (syndrome de Parinaud, 10 % des cas; à ne pas confondre avec le syndrome du même nom avec atteinte oculomotrice). Les signes sont : conjonctivite unilatérale folliculaire résistante aux antibiotiques locaux, chémosis, œdème palpébral et adénopathie prétragienne pouvant se compliquer de panuvéite avec décollement séreux rétinien, papillite et neurorétinite stellaire.
Toute plaie palpébrale ou orbitaire avec ou sans corps étranger ou infection de voisinage (ethmoïdite) pourra entraîner une cellulite orbitaire avec mise en jeu du pronostic visuel. En cas d'infection orbitaire, une antibiothérapie devra être mise en place en urgence et intensifiée ou modifiée en fonction de l'évolution clinique. Les signes de gravité sont : baisse d'acuité visuelle, mydriase, ophtalmoplégie.
Les infections oculaires d'origine fongique sont relativement rares en France en pédiatrie. La question d'une telle origine infectieuse doit être évoquée en cas de séjour en zone tropicale ou de terrain immunodéprimé. Il faut en revanche souligner le risque d'infection cornéenne fongique lié au port de lentilles de contact ou à une corticothérapie locale prolongée. Dans ces circonstances, l'isolement de l'agent infectieux avant traitement est indispensable par grattage cornéen et peut être complété par un examen en microscopie confocale; un traitement au cours d'une hospitalisation est indispensable (collyres antifongiques horaires, ± traitement systémique et injections intrastromales) [12, 13].
La collaboration entre les deux spécialistes est nécessaire dans la gestion des rétinochoroïdites qui doivent faire évoquer une toxoplasmose. La toxoplasmose constitue l'étiologie la plus fréquente des uvéites postérieures dans le monde [11]. Pour les formes acquises, c'est l'ophtalmologiste qui va être alerté par une atteinte de type de rétinochoroïdite : hyalite avec foyer blanchâtre à bords flous souvent profond, parfois associé à des lésions plus anciennes pigmentées. Il peut exister une uvéite antérieure granulomateuse. Des atteintes plus étendues, multifocales voire bilatérales, peuvent être observées. Les séquelles peuvent être multiples : cicatrices rétiniennes centrales avec baisse d'acuité visuelle, risque de néovascularisation secondaire, atteinte du nerf optique, décollement de rétine et phtyse [14-17]. La ponction de chambre antérieure, avec PCR sur l'humeur aqueuse et réalisation d'un coefficient de Desmond, permettra un diagnostic de certitude dans les formes douteuses acquises. L'apport de l'infectiologue permettra de confirmer le diagnostic et d'éliminer les diagnostics différentiels infectieux (tuberculose par exemple) ou rhumatologiques (Behçet) et d'aider à la prise en charge thérapeutique. Un bilan immunitaire sera nécessaire et assuré lors de la consultation pédiatrique. La mise en place du traitement est complexe compte tenu de la nécessité de deux antiparasitaires pour une durée prolongée de 6 semaines avec une toxicité hématologique, hépatique et rénale. Certaines formes sont récurrentes et devront nécessiter la mise en place d'un traitement préventif par l'infectiologue (par exemple Bactrim).
Habituellement bénigne, cette parasitose entraîne parfois des complications oculaires sévères, à type d'uvéites postérieures, atteignant particulièrement les enfants âgés de 2 à 7 ans. Elle serait responsable de 3 à 18 % des uvéites postérieures de l'enfant [18]. Deux nématodes (vers ronds) sont responsables de la toxocarose humaine : Toxocara canis, parasite du chien, et Toxocara cati, parasite du chat, qui ne deviennent adultes que chez le chien et dont les larves sont en impasse parasitaire chez l'homme. Outre la toxocarose oculaire qui touche principalement le grand enfant (7 ans), on rencontre deux autres formes de la maladie appelée larva migrans viscérale et larva migrans cutanée qui se rencontrent plus volontiers chez le grand nourrisson (2 ans).
Les manifestations oculaires sont parfois l'unique signe d'appel de la maladie : granulome rétinien postérieur qui en est la manifestation la plus fréquente (lésion pseudo-tumorale, parfois bilatérale responsable de phénomènes tractionnels rétiniens), hyalite, uvéite granulomateuse d'origine immuno-allergique, neurorétinite subaiguë [19]. Le traitement devra associer un antiparasitaire par voie systémique (Zentel) pendant 8 à 15 jours à une corticothérapie orale devant le risque de majoration des signes inflammatoires par lyse parasitaire.
Devant la découverte d'une uvéite, le pédiatre sera sollicité à la recherche d'une atteinte infectieuse ou inflammatoire. Un bilan complémentaire sera nécessaire en collaboration avec les rhumatologues (arthrite juvénile idiopathique, etc.), les infectiologues (tuberculose, bartonellose, rickettsiose, maladie de Lyme, hépatites, virus de l'immunodéficience humaine, toxoplasmose, toxocarose, etc.) et les pneumologues pédiatres (sarcoïdoses) (voir plus loin et chapitre 14).
L’infectiologue a besoin de l’ophtalmologiste pour :
bilan des foetopathies TORCH ;
kératoconjonctivites du nouveau-né ;
herpès et varicelle ;
septicémies.
L’ophtalmologiste a besoin de l’infectiologue pour :
herpès et varicelle avec traitement systémique ;
conjonctivite avec retentissement systémique ;
abcès cornéens fongiques ;
toxoplasmose et toxocarose ;
granulomatoses (tuberculose) et autres uvéites infectieuses.
Remerciements au Pr Marc Labetoulle (Paris).
[1] Bodaghi B Les uvéites virales Journal Français d'Ophtalmologie: ( 2004 ) : 27: 528-537
[2] Brezin AP, Thulliez P, Cisneros B Lymphocytic choriomeningitis virus chorioretinitis mimicking ocular toxoplasmosis in two otherwise normal children Am J Ophthalmol: ( 2000 ) : 130: 245-247
[3] Mets MB, Barton LL, Khan AS, Ksiazek TG Lymphocytic choriomeningitis virus : an underdiagnosed cause of congenital chorioretinitis Am J Ophthalmol: ( 2000 ) : 130: 209-215
[4] Teoh DL, Reynolds S Diagnosis and management of pediatric conjonctivitis Pediatr Emerg Care: ( 2003 ) : 19: 48-55
[5] Darville T Chlamydia trachomatis infections in neonates and young children Semin Pediatr Infect Dis: ( 2005 ) : 16: 235-244
[6] MacDonald N, Mailman T Gonoccocal infections in newborns and in adolescents Adv Exp Med Biol: ( 2008 ) : 609: 108-130
[7] Serna-Ojeda JC, Ramirez-Miranda A, Navas A Herpes Simplex Virus disease of the anterior segment in children Cornea: ( 2015 ) : 34: 68-71
[8] Miserocchi E, Waheed NK Visual outcome in herpes simplex virus and varicella zoster virus uveitis Ophtalmology: ( 2002 ) : 109: 1532-1537
[9] Naseri A, Good WV, Cunningham ET Herpes zoster virus sclerokeratitis and anterior uveitis in a child following varicella vaccination Am J Ophthalmol: ( 2003 ) : 135: 415-417
[10] Rousseau A, Labetoulle M Herpès et varicelle-zona chez l'enfant : quelles particularités? Réalités Ophtalmologiques: ( 2015 ) 221-222
[11] Invernizzi A, Mameli C, Giacomet V Herpetic acute anterior uveitis complicated by retinal vasculitis in an immunocompetent child Can J Ophthalmol: ( 2013 ) : 48: 171-172
[12] Kalkanci A, Ozdek S Ocular fungal infection Curr Eye Res: ( 2011 ) : 36: 179-189
[13] Bourcier T, Sauer A Kératites fongiques J Fr Ophtalmol: ( 2011 ) : 34: 563-567
[14] Henderly DE, Genstler AJ, Smith RE, Rao NA Changing patterns of uveitis Am J Ophthalmol: ( 1987 ) : 103: 131-136
[15] Kodjikian L, Wallon M Ocular manifestations in congenital toxoplasmosis Graefes Arch Clin Exp Ophtalmol: ( 2006 ) : 244: 14-21
[16] Wallon M, Kodjikian L Long-term ocular prognosis in 327 children with congenital toxoplasmosis Pediatrics: ( 2004 ) : 113: 1567-1572
[17] Vutova K, Peicheva Z, Popova A Congenital toxoplasmosis : eye manifestations in infants and children Ann Trop Paediatr: ( 2002 ) : 22: 213-218
[18] Pawlowski Z Toxocariasis in humans : clinical expression and treatment dilemma J Helminthol: ( 2001 ) : 75: 299-305
[19] Sauer A, Candolfi E, Speeg-Schatz E, Bourcier T Toxocarose oculaire : de la clinique au diagnostic Réflexions Ophtalmologiques: ( 2009 ) : 14: 262-264
A. -L. Jurquet, C. Benso-Layoun
La rhumatologie pédiatrique et l'ophtalmologie ont des liens étroits du fait de la présence d'atteinte ophtalmologique éventuelle dans la plupart des pathologies inflammatoires et auto-immunes concernées.
Lors d'un diagnostic en rhumatologie pédiatrique, une recherche de signes ophtalmologiques qui auraient pu passer inaperçus auprès du patient et de sa famille est très souvent demandée. De plus, en cas de normalité de l'examen initial, il pourra être nécessaire dans certains cas de pratiquer des examens ophtalmologiques réguliers afin de détecter une atteinte secondaire.
A contrario, les ophtalmologistes font appel aux rhumatologues devant un diagnostic de pathologie oculaire inflammatoire, soit lors de leur prise en charge initiale soit au cours de l'évolution dans le but de détecter une pathologie inflammatoire ou auto-immune non exclusivement ophtalmologique.
Une fois passée l'étape du diagnostic étiologique, la collaboration va être également effective pour l'initiation de certains traitements, leur indication selon les cas et leur surveillance.
C'est la pathologie inflammatoire la plus fréquente de l'enfant concernant, selon une étude épidémiologique, plus de 59 000 patients en Europe en 2010 [1]. Le risque de survenue d'une uvéite, classiquement sur un œil blanc, calme et indolore, est important et maximal pendant les 4 voire 5 premières années du suivi de cette pathologie chronique. Une étude récente allemande [2] estime la prévalence de l'uvéite associée à l'AJI à 11 % en 2013 (avec survenue de 23 % de complications). Il est donc nécessaire de pratiquer un examen ophtalmologique à la lampe à fente régulier, à une fréquence déterminée selon le type d'AJI, le sexe et la présence ou non de facteurs antinucléaires. Il existe aussi des uvéites inaugurales d'AJI dans environ 10 % des cas. De nombreux patients sont alors suivis en ophtalmologie de ville, mais nécessitent parfois l'expertise des ophtalmologistes du centre hospitalo-universitaire spécialisés dans le domaine.
En dehors des uvéites associées à l'AJI, nous suivons conjointement une file active de patients atteints d'uvéites idiopathiques nécessitant :
- – un bilan étiologique complet guidé par les caractéristiques de l'examen ophtalmologique (avec recours parfois à des plateaux techniques d'imagerie et anesthésie pédiatriques notamment pour les ponctions de chambre antérieure utiles pour les diagnostics différentiels infectieux);
- – un traitement parfois systémique;
- – un suivi ultérieur régulier de la tolérance des traitements mais aussi du dépistage de potentiels signes extra-ophtalmologiques pouvant faire porter un diagnostic secondaire de pathologie générale.
Aussi, des traitements par corticoïdes (intraveineux, per os), par immunosuppresseurs (azathioprine, méthotrexate), ou par biothérapies (anti-tumor necrosis factor [anti-TNF]) sont régulièrement discutés, initiés, ajustés et surveillés.
Les dermatomyosites juvéniles, les lupus érythémateux disséminés ou autre connectivites, la sarcoïdose (syndrome de Blau à début pédiatrique ou forme plus tardive se rapprochant de la forme adulte) comportent également des atteintes ophtalmologiques primordíales à détecter. Dans le cadre des connectivites, l'autorisation et le suivi d'un traitement par antipaludéen de synthèse par les ophtalmologistes sont nécessaires.
La maladie de Behçet est particulièrement concernée et la présence d'une uvéite fait partie des critères diagnostiques.
Au sein de ce domaine en pleine expansion, citons :
- – les pathologies associées à la cryopyrine ou cryopyrin-associated periodic syndromes (CAPS) : syndrome Chronic Infantile Neurological Cutaneous and Articular (CINCA), syndrome de Muckle-Wells;
- – les pathologies associées au récepteur du TNF (TNF-receptor associated periodic syndrome [TRAPS]);
- – le déficit en mévalonate kinase (ou syndrome hyper-IgD);
- – la fièvre méditerranéenne familiale.
Il est important de rechercher des signes en rapport avec ces entités au niveau ophtalmologique, lors de la prise en charge de patients atteints de maladie de Marfan ou de maladie d'Ehlers-Danlos.
- – Renforcement des interactions en formalisant, par exemple, des consultations communes au sein de la même unité de temps et de lieu pour des pathologies comme les uvéites, pour lesquelles il a été montré chez l'adulte que la coopération ophtalmologiste-interniste accroissait les chances de diagnostic étiologique [3].
- – Développement de thématiques de recherche commune, comme l'étude récente testant un anti-TNF (adalimumab) sur tout le territoire national pour le traitement d'uvéites sévères dans l'AJI (protocole ADJUVITE). Éducation thérapeutique du patient avec programme en cours d'élaboration pour l'AJI, un volet concernant l'atteinte ophtalmologique pouvant être développé.
Pathologies concernées :
-
AJI ;
uvéites idiopathiques ;
connectivites ou autres pathologies inflammatoires ;
vascularites : la maladie de Behçet est particulièrement concernée, et la présence d’une uvéite fait partie des critères diagnostiques ;
maladies auto-immunes : pathologies associées à la cryopyrine, déficit en mévalonate kinase (ou syndrome hyper-IgD) et fièvre méditerranéenne familiale ;
pathologies du tissu conjonctif : maladie de Marfan ou de maladie Ehlers-Danlos.
Perspectives nombreuses :
-
renforcement des interactions en formalisant des consultations communes ;
développement de thématiques de recherche commune (anti-TNF : adalimumab) ;
programme d’éducation thérapeutique (AJI).
Au total, l'exposé des grandes lignes de notre activité en rhumatologie pédiatrique tend à mettre en lumière la collaboration quotidienne entre pédiatres et ophtalmologistes impliqués dans la prise en charge d'enfants. Nous avons besoin les uns des autres à toutes les étapes de la prise en charge, souvent multidisciplinaire, de nos patients. L'aide apportée par des ophtalmologistes rompus au difficile exercice parfois de l'examen de très jeunes enfants est pour les rhumatologues primordiale.
[1] Thierry S, Fautrel B, Lemelle I, et al. Prevalence and incidence of juvenile idiopathic arthritis : a systematic review. Joint Bone Spine 2014 ; 81 : 112-7.
[2] Tappeiner C, Klotsche J, Scenck S, et al. Temporal change in prevalence and complications of uveitis associated with juvenile idiopathic arthritis : data from a crosssectional analysis of a prospective nationwide study. Clin Exp Rheumatol 2015 ; 33 :936-44.
[3] Le Scanff J, Seve P, Kodjikian L, et al. Interest of an internist’s consultation in uveitis.Comparative study in 66 cases. Rev Med Interne 2006 ; 27 : 671-8.
C. Barraud, B. Chabrol , F. Audic, E. Zanin
De nombreuses pathologies peuvent présenter une atteinte conjointe ophtalmologique et neurologique. L'atteinte ophtalmologique constitue parfois le premier signe d'appel et aide au diagnostic étiologique ou, inversement, elle est recherchée dans le cadre d'un bilan diagnostique d'une affection neurologique. Une collaboration entre les deux spécialités est primordiale.
Il faut en premier lieu éliminer un processus expansif intracrânien, notamment une tumeur de la fosse postérieure. Une imagerie cérébrale sera donc réalisée à cet effet après examen clinique complet.
L'HTIC idiopathique, souvent associée à un contexte d'obésité en particulier chez les jeunes filles, est une cause fréquente d'œdème papillaire le plus souvent bilatéral [1, 2]. La plupart de ces patients sont adressés aux neuropédiatres par les ophtalmologistes. Le tableau clinique s'accompagne le plus souvent de céphalées et/ou de vomissements. Une imagerie cérébrale type angio-IRM est indispensable dans ce cadre pour éliminer les diagnostics différentiels (tumeur cérébrale, thrombophlébite cérébrale, etc.). La confirmation du diagnostic est faite par la mesure de pression du liquide céphalorachidien (> 15 cmHO si indice de masse corporelle normal; > 20 cmHO chez le patient obèse). Le traitement comporte de l'acétalozamide (Diamox) et une prise en charge diététique pour perte de poids chez les patients obèses ainsi qu'un suivi pluridisciplinaire rapproché. En cas de pronostic visuel engagé (baisse de la vision, atrophie optique) malgré une bonne compliance au traitement, une intervention neurochirurgicale est parfois nécessaire, et doit toujours être discutée de façon pluridisciplinaire.
Face à une hypoplasie bilatérale des nerfs optiques chez un nourrisson qui présente un trouble du comportement visuel, il faudra rechercher des anomalies de la ligne médiane. Le bilan recherchera également des associations systémiques et tératogéniques.
La compression tumorale du nerf optique telle qu'on peut le voir dans les cas de craniopharyngiome (tumeur épithéliale bénigne se développant dans la région sellaire et suprasellaire) peut mener à une atrophie optique uni- ou bilatérale tout comme les gliomes des voies optiques.
Toute neuropathie optique inflammatoire peut également aboutir à une atrophie optique. On peut également retrouver des atrophies optiques acquises dans le cadre de pathologies neurologiques diverses (séquelles d'anoxie périnatale, de méningite et/ou d'encéphalite, de traumatisme crânien ou encore d'hydrocéphalie) et dans de rares cas telles que certaines maladies métaboliques dégénératives.
Le tableau clinique comporte une baisse d'acuité visuelle brutale uni- ou bilatérale avec le plus souvent douleur à la mobilisation des globes oculaires et parfois perte de la vision des couleurs. Les causes neurologiques à évoquer sont la sclérose en plaques en premier lieu [3] et son apparenté, la neuromyélite optique (NMO) de Devic qui reste cependant rare chez l'enfant. Ces maladies répondent à des critères diagnostiques très précis. Un bilan paraclinique est alors nécessaire avec imagerie par résonance magnétique (IRM) cérébromédullaire à la recherche de plaques de démyélinisation de la substance blanche et ponction lombaire à la recherche d'une synthèse intrathécale ou la positivité d'anticorps tels que les anti-NMO. Un traitement par bolus de corticoïdes permet une résolution des symptômes le plus souvent en quelques jours.
Une POM du VI doit faire évoquer en premier lieu une HTIC (notamment due à une tumeur de la fosse postérieure), bien que l'atteinte du VI n'ait pas de valeur localisatrice. Il existe aussi des atteintes inflammatoires (Miller-Fisher) ou par infection locorégionale.
La POM III globale correspond le plus souvent à une atteinte inflammatoire avec hypertrophie du nerf à l'imagerie cérébrale. Un traitement par corticothérapie courte per os permet en général une résolution totale des symptômes.
À noter que devant une paralysie de la verticalité (syndrome prétectal de Parinaud), il faut évoquer avant tout une lésion de la partie postérosupérieure du tronc cérébral (le plus souvent tumorale) et, en l'absence de lésion, des pathologies métaboliques très rares telles que la maladie de Niemann-Pick de type C ou encore la maladie de Gaucher type II ou III, mais dans ce cas, la POM n'est jamais isolée [4].
Le ptosis fluctuant dans la journée associé ou non à une diplopie et/ou une ophtalmoplégie doit faire évoquer la possibilité d'une myasthénie [5]. Il s'agit d'une affection de la jonction neuromusculaire qui peut être isolée. Elle peut être auto-immune ou congénitale. Le caractère fluctuant, aggravé par l'effort est très évocateur. Le diagnostic sera confirmé par une exploration neurophysiologique à la recherche d'un décrément. Un test aux anticholinestérasiques peut parfois s'avérer utile.
Une apraxie oculomotrice peut révéler des atteintes cérébelleuses ou pontocérebelleuses et nécessite une imagerie cérébrale et la recherche de signes associés : atteinte rétinienne, rénale, hépatique ou neuropathie périphérique.
Le nystagmus du nourrisson est un motif fréquent de collaboration neuro-ophtalmologique [6]. En effet, même s'il est le plus souvent bénin, un bilan ophtalmologique (avec électrophysiologie) est toujours indispensable en première intention chez ces enfants à la recherche d'une rétinite pigmentaire, d'une cataracte congénitale, d'une maculopathie congénitale, etc. Dans tous les cas, il est important de réaliser un examen neurologique à la recherche de signes associés (cachexie, hypotonie, etc.). Une IRM cérébro-orbitaire sera réalisée secondairement pour rechercher principalement une anomalie tumorale ou malformative, plus rarement une anomalie de la myélinisation, ainsi que pour vérifier les centres de la stabilité du regard (réticulée du tronc cérébral, noyaux vestibulaires, colliculi supérieurs).
Un examen clinique complet est indispensable à la recherche notamment d'une ataxie, de troubles du comportement/sommeil ou de myoclonies faisant évoquer un syndrome opsomyoclonique dont l'étiologie la plus fréquente est un neuroblastome (syndrome paranéoplasique) chez le nourrisson et le jeune enfant. Il convient donc de réaliser un bilan d'imagerie adapté (IRM corps entier) ainsi que le dosage des catécholamines urinaires. Un traitement prolongé par dexaméthasone en bolus et la prise en charge de la tumeur si cela est possible améliorent le pronostic de ces enfants, bien que l'atteinte neurologique évolue souvent pour son propre compte.
Lorsque le bilan ophtalmologique ne retrouve pas de cause réfractive, oculomotrice ou organique pouvant expliquer les difficultés scolaires, on pourra évoquer un trouble praxique ou une atteinte cognitive.
Le plus souvent, les nourrissons sont vus aux urgences pédiatriques pour des tableaux d'hypotonie/troubles de la vigilance, malaises ou convulsions. L'imagerie cérébrale permet de mettre en évidence un hématome sous-dural uni- ou plurifocal. Au niveau ophtalmologique, les hémorragies rétiniennes (HR) sont quasi pathognomoniques du syndrome du bébé secoué quand elles sont multiples, profuses ou éclaboussant la rétine jusqu'à sa périphérie, avec parfois rétinoschisis hémorragique et/ou pli rétinien périmaculaire (HR de type 3). La coexistence d'une histoire clinique absente, incohérente ou incompatible, de lésions cérébrales et ophtalmologiques fait porter le diagnostic de traumatisme crânien infligé par secousses [7].
La neurofibromatose de type 1 (NF1) est la phacomatose la plus fréquente. La transmission est autosomique dominante, il s'agit d'une mutation du gène NF1 suppresseur de tumeur aboutissant à un dysfonctionnement du tissu ectodermique embryonnaire formant la peau, le système nerveux et l'œil expliquant l'atteinte possible à ces trois niveaux. Les critères diagnostiques sont la présence de taches café au lait, de lentigines, de neurofibromes, de gliome des voies optiques, de lésions osseuses caractéristiques, d'antécédents de NF1 au premier degré et de nodules de Lisch (hamartomes iriens asymptomatiques) à la lampe à fente. Les nodules de Lisch sont caractéristiques de la maladie (tout comme les anomalies choroïdiennes en infrarouge récemment décrites [8-10]). L'important est de surveiller le niveau d'acuité visuelle car c'est lui qui conditionne la mise en route d'un traitement et non l'existence d'un gliome (de nombreux gliomes peuvent être asymptomatiques ou disparaître spontanément).
Ils sont présents dans 50 % des cas. Les signes neurologiques comprennent des crises convulsives, un retard mental et des tubercules intracérébraux.
- – Anneaux de Kayser-Fleischer à la lampe à fente (formations arrondies de couleur jaune verdâtre présentes à la périphérie de l'iris des yeux dues à l'accumulation de sels de cuivre dans le sang) dans la maladie de Wilson.
- – Luxation du cristallin dans l'homocystinurie.
- – Opacités cornéennes avec parfois glaucomes ou rétinopathies associés dans les maladies de surcharge telles que certaines mucopolysaccharidoses.
- – Rétinite pigmentaire dans certaines maladies mitochondriales qui, associée à un ptosis et une ophtalmoplégie, fait évoquer un syndrome de Kearns-Sayre.
- – Tache maculaire rouge cerise notamment dans les gangliosidoses GM2 (maladie de Tay-Sachs).
Ce que le neuropédiatre apporte à l’ophtalmologiste :
-
oedème papillaire : HTIC tumorale ou idiopathique ;
atrophie optique : étiologie tumorale ou inflammatoire ;
neuropathie optique rétrobulbaire : sclérose en plaques, syndrome de Devic ;
paralysie oculomotrice (VI, III) : tumeur fosse postérieure, HTIC ;
ptosis, ophtalmoplégie : myasthénie ;
nystagmus : causes neurologiques ;
opsoclonies : syndrome opsomyoclonique, neuroblastome.
Ce que l’ophtalmologiste apporte au neuropédiatre :
-
syndrome du bébé secoué : hémorragies rétiniennes ;
neurofibromatose : nodules de Lisch ;
bilan d’extension des maladies métaboliques.
[1] Brara SM Pediatric idiopathic intracranial hypertension and extreme childhood obesity J Pediatr: ( 2012 ) : 161: 602-607
[2] Salpietro V Pediatric idiopathic intracranial hypertension and extreme childhood obesity : a role for weight gain. J Pediatr: ( 2013 ) : 162: 1084-
[3] Heussinger N, Kontopantelis E, Gburek-Augustat J Oligoclonal bands predict multiple sclerosis in children with optic neuritis Ann Neurol: ( 2015 ) : 77: 1076-1082
[4] Burztyn J, Mayer M Neuroophtalmologie Neurologie pédiatrique: Médecine Sciences/Flammarion ( 2010 ) 920-929
[5] Liew WKM, Kang PB Update on juvenile myasthenia gravis Curr Opin Pediatr: ( 2013 ) : 25: 694-700
[6] Denis D, Girard N, Toesca E MRI in congenital nystagmus J Fr Ophtalmol: ( 2010 ) : 33: 189-205
[7] Haute Autorité de Santé (HAS) Syndrome du bébé secoué. Rapport d'orientation de la commission d'audition HAS : -
[8] Parrozzani R In vivo detection of choroidal abnormalities related to NF1 : feasibility and comparison with standard NIH diagnostic criteria in pediatric patients IOVS: ( 2015 ) 6036-6042
[9] Prada CE, Hufnagel RB, Hummel TR The use of magnetic resonance imaging screening for optic pathway gliomas in children with neurofibromatosis type 1 J Pediatr: ( 2015 ) : 167: 851-856
[10] Pinson S, Créange A, Barbarot S Recommendations for the treatment of neurofibromatosis type 1 J Fr Ophtalmol: ( 2002 ) : 25: 423-433
M. -C. Koeppel , E. Zanin, M. Callet, S. Mallet, D. Denis
Dermatopédiatres et ophtalmopédiatres sont en étroite collaboration au niveau du diagnostic et de la prise en charge des pathologies oculocutanées, qu'elles soient congénitales ou acquises. L'origine embryologique commune neuro-ectodermique des deux structures, mais aussi la continuité palpébroconjonctivale des tissus ou l'exposition identique aux agressions extérieures en tant qu'épithélium de surface (micro-organismes, allergènes ou facteurs environnementaux) expliquent la fréquence des pathologies oculocutanées et leurs intrications.
La peau étant plus visible et accessible que l'œil, le motif initial de consultation de l'enfant est la présence de lésions cutanées, l'ophtalmologiste étant sollicité secondairement à la recherche d'atteinte spécifique.
- – Orgelet : furoncle du cil correspondant à une nécrose staphylococcique aiguë d'un bulbe pileux.
- – Chalazion : nodule sous-cutané intratarsal correspondant à un granulome de résorption d'une glande de Meibomius.
- – Impétigo : dermatose contagieuse à staphylocoque ou streptocoque entraînant des lésions croÛteuses ou bulleuses, fréquente chez les enfants en bas âge, à début péri-orificiel et dissémination secondaire.
- – Cellulite préseptale à Haemophilus influenzae.
- – Peau : les formes cliniques sont nombreuses.
- – Œil : chancre palpébral, conjonctivite, phlyctène conjonctival, sclérite nécrosante et non nécrosante, kératite interstitielle, uvéite antérieure granulomateuse, choroïdite multifocale avec tubercules de Bouchut, granulome choroïdien, vascularite, périphlébite, maladie de Eales, choroïdite serpigineuse, épithéliopathie en plaques, œdème papillaire, papillite, neuropathie optique et neurorétinite.
- – Peau : trois phases primaire (chancre d'inoculation avec adénopathie satellite), secondaire (roséole, syphilides, atteinte des muqueuses, alopécie) et tertiaire (gommes, nodules).
- – Œil (au cours des phases secondaire et tertiaire) : conjonctivite nodulaire, sclérite, kératite interstitielle, uvéite, choriorétinite multifocale, nécrose rétinienne aiguë, vascularite, neuropathie optique, paralysie oculomotrice dans le cadre d'une méningite, signe Argyll-Robertson en cas d'atteinte tertiaire avec tabès.
- – Peau : contamination par griffure avec apparition 5 à 10 jours après d'une papule transitoire et d'une adénite dans un contexte fébrile.
- – Œil :
10 % d’inoculation conjonctivale par pelage contaminé (syndrome de Parinaud) avec une conjonctivite unilatérale folliculaire résistante aux antibiotiques locaux, chémosis, oedème palpébral et adénopathie prétragienne ;
panuvéite avec décollement séreux rétinien et papillite ;
neurorétinite stellaire.
- – Peau : 3 phases primaire (erythema chronicum migrans), secondaire (lymphocytome cutané bénin : nodule violacé ferme) et tertiaire (acrodermite atrophiante).
- – Œil (75 % des cas lors de la phase tardive de la maladie) :
-
primaire : conjonctivite bilatérale ;
secondaire ou tertiaire : épisclérite, sclérite, uvéite, kératite intersticielle, myosite, paralysie oculomotrice, vascularite rétinienne, occlusion veineuse, neuropathie optique ischémique antérieure, papillite, endophtalmie.
- – Peau : trois types (lèpre tuberculoïde, borderline et lèpre lépromateuse).
- – Œil (en cas d'atteinte généralisée) :
-
lépromes des paupières avec troubles de la statique palpébrale, lépromes conjonctivaux, kératite interstitielle, hypertrophie des nerfs cornéens puis anesthésie cornéenne ;
lagophtalmie sévère par troubles de la statique palpébrale ;
uvéite micronodulaire synéchiante chronique.
- – Peau : érythème, œdème avec vésicules ou ulcérations possibles au niveau péri-oculaire.
- – Œil : conjonctivite, kératite dendritique, kératite interstitielle, endothélite, uvéite avec hypertonie oculaire.
- – Cas particulier du syndrome de Kaposi-Juliusberg : dissémination brutale du virus herpétique sur une dermatose préexistante (dermatite atopique par exemple) responsable d'une altération de l'état général, d'une fièvre, d'une éruption diffuse vésiculeuse, sévère au niveau du visage avec atteinte oculaire possible.
- – Contamination in utero par dissémination hématogène transplacentaire en cas de primo-infection maternelle avec virémie élevée ou par voie ascendante en cas de rupture prématurée des membranes. Risque d'avortements spontanés, accouchements prématurés, RCIU, lésions cutanées in utero, microcéphalie, calcification intracérébrale, hydrocéphalie, hémorragie cérébrale, choriorétinite, microphtalmie, cataracte, aplasie maculaire ou papillaire, chorio-amniotite.
- – Contamination per-partum lors du passage dans la filière génitale, risque maximal en cas de primo-infection récente (40 à 70 % d'atteinte). Survient avant le 28 jour. La forme bénigne survient vers le 6 jour et associe une atteinte cutanée, buccale et oculaire. Les formes graves peuvent être localisées au système nerveux central avec une méningo-encéphalite ou disséminées avec une atteinte hépatique, pulmonaire et neurologique.
- – Contamination post-natale rare.
- – Peau : douleur puis érythème, œdème et éruption de vésicules ombiliquées au niveau du territoire du V1.
- – Œil : conjonctivite folliculaire, kératite dendritique, kératite interstitielle, uvéite (récidivante).
- – Peau et paupières : sarcome de Kaposi, à herpèsvirus humain de type 8, entraînant des lésions violacées avec atteinte du bord libre.
- – Œil : Rétinopathie liée au VIH (micro-angiopathie), choriorétinites par infections opportunistes (CMV, VZV, toxoplasmose, tuberculose, cryptoccoque, candidoses, etc.).
- – Molluscum contagiosum : infection à poxvirus, contagieux chez l'enfant, dissémination par grattage, peut être responsable d'une kératoconjonctivite folliculaire.
- – Verrues palpébrales difficiles à traiter étant donné la localisation anatomique.
Pédiculoses.
Onchocercose (filaire transmise par une morsure de mouche noire, pathologie endémique de certaines régions d'Afrique et d'Amérique centrale) :
- – peau : multiples petites papules dispersées, prurigineuses, siégeant sur les fesses et les membres; nodules sous-cutanés (région rétro-auriculaire ++);
- – œil : microfilaires visibles en chambre antérieure, kératite avec opacités stromales, kératite sclérosante, choriorétinite.
Syndrome adéno-cutanéo-muqueux aigu fébrile lié à une panvascularite systémique des artères de gros et moyen calibre. Une étiologie infectieuse est suspectée comme élément déclenchant. Le pronostic est dominé par l'atteinte cardiaque et la survenue d'anévrismes coronariens. La prise en charge thérapeutique doit être faite en urgence pour les prévenir.
- – Peau :
-
pharyngite érythématopultacée puis chéilite et stomatite ;
atteinte des extrémités avec érythème, oedème des mains et des pieds suivis d’une phase de desquamation en doigts de gant ;
exanthème polymorphe non prurigineux du torse au 3e jour.
- – Œil (90 % des cas) :
-
conjonctivite bilatérale non purulente apparaissant dans la semaine suivant la fièvre et persistant 15 jours avec régression sans traitement ;
uvéite antérieure non granulomateuse.
- – Peau : eczéma des paupières, signe de Dennie-Morgan.
- – Œil :
-
conjonctivite papillaire avec chémosis ;
kératoconjonctivite vernale (terrain : garçon de 10 ans, recrudescence estivale) : papilles géantes, pseudo-ptosis, sécrétions mucineuses, bourrelet limbique, grains de Trantas, kératite, ulcère vernal en phase aiguë, complications fréquentes (kératocône, taie cornéenne, astigmatisme irrégulier, amblyopie, complications iatrogéniques de la corticothérapie locale – glaucome, cataracte – en phase chronique).
- – Peau : érythème télengiectasique de la face avec bouffées vasomotrices.
- – Œil : dysfonctionnement meibomien, blépharite, chalazion, syndrome sec, kératite ponctuée superficielle, kératite interstitielle, conjonctivite phlycténulaire, infiltrats stromaux inférieurs avec néovascularisation cornéenne, astigmatisme irrégulier, taie cornéenne, amblyopie.
- – Peau : dermatose érythématosquameuse, l'atteinte du visage est rare sauf chez l'enfant avec une localisation palpébrale de prédilection.
- – Œil : blépharite, kératoconjonctivite, syndrome sec, uvéite antérieure non granulomateuse très rare, complications iatrogènes des dermocorticoïdes ou de la PUVA-thérapie (de moins en moins utilisée).
Cet hamartome mélanoblastique du territoire trigéminal congénital non héréditaire et unilatéral est plus fréquent chez les patients asiatiques.
- – Peau : mélanose cutanée de teinte bleu ardoisé unilatérale située sur une branche du V.
- – Œil : atteinte ophtalmologique pouvant être isolée (melanosis oculi), atteinte de la sclère dans 100 % des cas, conjonctive et iris dans 50 % des cas, anneau scléral postérieur rare, complications à titre de glaucome et mélanome (surtout chez les patients peu pigmentés).
Urgences dermatologiques mettant en jeu le pronostic vital. Principaux médicaments en cause : sulfamides, anticonvulsivants, anti-inflammatoire non stéroïdien (AINS), allopurinol, etc.
- – Peau :
-
Stevens-Johnson : lésions en cocarde, macules purpuriques ou ardoisées étendues et bulleuses, décollement cutané inférieur à 10 % , atteinte des muqueuses ;
Lyell : prodromes pseudo-grippaux suivis d’une éruption fébrile scarlatiniforme débutant au visage puis s’étendant au reste du corps associée à un oedème palmo-plantaire, décollement cutané en linge mouillé > 30 % , signe de Nikolski, atteinte des muqueuses dans 95 % des cas.
- – Œil : conjonctivite bilatérale pseudo-membraneuse avec des paupières adhérentes aboutissant à des symblépharons, entropions, trichiasis, syndrome sec, exposition cornéenne, kératite, ulcère. Séquelles chez plus de 50 % des survivants : obstructions des méats lacrymaux, pannus cornéen, infiltration lymphocytaire des glandes lacrymales, leucome cornéen.
Ichtyoses : groupe hétérogène d'anomalies de la kératinisation résultant d'une rétention épidermique ou d'une prolifération épidermique. Atteinte palpébrale avec ectropion, symblépharon, obstruction des méats lacrymaux, diastasis oculopalpébral avec kératinisation cornéenne, lagophtalmie.
- – Peau : ichtyose noire ou nigricans.
- – Œil : 50 % d'opacités cornéennes.
- – Transmission autosomique récessive, génétiquement hétérogène.
- – Peau : bébé collodion à la naissance puis érythrodermie sèche.
- – Œil : ectropion.
- – Autosomique récessif.
- – Peau : érythrodermie ichtyosiforme dès la naissance, puis érythrokératodermie avec lésions verruqueuses prédominant au visage, hyperkératose palmoplantaire, alopécie, onychodystrophie, carcinomes cutanés, infections parfois sévères.
- – Œil : atteinte cornéenne dans 84 % des cas secondaire à une insuffisance limbique grave.
Trouble de la mélanogenèse, incidence 1/15 000, deux types tyrosinase négatif ou positif. Le type tyrosinase négatif réalise un tableau complet, les autres types sont des variants phénotypiques.
- – Peau : cheveux blancs, peau blanche ou rosée, absence de bronzage, risque de carcinomes multiples et de mélanomes.
- – Œil : iris diaphane, transilluminable, photophobie, nystagmus pendulaire apparaissant entre 6 et 12 mois, strabisme, amblyopie, astigmatisme, fond d'œil hypopigmenté avec hypoplasie fovéolaire, réponses croisées aux potentiels évoqués visuels (PEV) témoignant d'une décussation quasi totale des fibres ganglionnaires au niveau chiasmatique.
Maladie neuro-ectodermique, à transmission dominante liée à l'X, avec létalité masculine et expressivité variable entraînant une atteinte cutanée, dentaire, squelettique, ophtalmologique et neurologique (épilepsie, retard mental).
- – Peau : aspect de dermatose pigmentaire en éclaboussures avec un début à la naissance et une évolution en trois phases successives ou intriquées (éruption vésiculeuse ou bulleuse, suivant les lignes de Blaschko, prédominant au niveau des membres inférieurs, puis stade verruqueux hyperkératosique ou lichénoïde, suivi par une hyperpigmentation s'effaçant progressivement).
- – Œil : atteinte dans 40 % des cas justifiant une surveillance étroite du fond d'œil dès la première année de vie. Vascularite rétinienne périphérique ischémique avec prolifération néovasculaire, altération de l'épithélium maculaire, dystrophie rétinienne, cataracte, microphtalmie, atrophie optique, strabisme, nystagmus.
Pathologie neurocutanée multisystémique touchant la peau, le squelette, l'œil et le système nerveux central. Sa présentation très variable serait due à un mosaïcisme cutané d'une anomalie génétique.
- – Peau : lésions hypopigmentées le long des lignes de Blaschko.
- – Œil : cataracte, strabisme, nystagmus, dystrophie rétinienne.
Génodermatose rare autosomique récessive favorisée par la consanguinité. Dysfonctionnement du système de réparation des anomalies de l'acide désoxyribonucléique (ADN) engendrées par les ultraviolets (UV).
- – Peau : absence de lésions à la naissance puis apparition progressive, lésions prédominant aux zones photo-exposées, aspect poïkilodermique de la peau, lentigines, kératose actinique, kératoacanthome, carcinomes basocellulaires et épidermoïdes de survenue précoce, mélanomes.
- – Œil : atteinte cutanée des paupières avec ectropion et tumeurs, kératite, carcinome épidermoïde du limbe, carcinomes et mélanomes conjonctivaux.
- – Peau : érosions et ulcérations cutanéomuqueuses diffuses d'apparition précoce évoluant vers des cicatrices dystrophiques, alopécie, absence d'ongles, syndactylies, parfois sténoses œsophagienne et anale, risques de surinfection cutanée, carcinomes épidermoïdes fréquents chez l'adulte jeune.
- – Œil : 75 % de lésions epithéliales cornéoconjonctivales (érosions, ulcérations), symblépharons, ectropions, carcinome épidermoïde palpébral fréquent.
Malformation vasculaire fréquente du nourrisson. Elle est discrète à la naissance puis subit une phase de prolifération pendant 9 mois avec un retentissement visuel rapide : amputation de l'axe visuel et astigmatisme responsables d'une amblyopie fonctionnelle.
Pathologie non héréditaire d'étiologie inconnue associant une malformation capillaire faciale à une atteinte ophtalmologique et neurologique (extension leptoméningée avec retard psychomoteur, épilepsie).
- – Peau : angiome plan présent à la naissance dans la région frontale et palpébrale supérieure (territoire V1) pouvant s'étendre dans les territoires V2 (maxillaire) et V3 (mandibulaire). L'atteinte peut être uni- ou bilatérale.
- – Œil : glaucome, hémangiome choroïdien diffus pouvant entraîner un œdème maculaire cystoïde et un décollement de rétine exsudatif.
Maladie vasculaire congénitale localisée ou généralisée. Les signes cutanés sont associés dans 50 % des cas à des signes extracutanés (macrocéphalie, etc.).
- – Peau : atteinte précoce, réseau vasculaire bleu-violet réticulé asymétrique avec télangiectasies, angiome stellaire, ectasie veineuse voire ulcération et atrophie. Touche préférentiellement les membres et rarement le visage.
- – Œil : glaucome et décollement de rétine exsudatif en cas d'atteinte du visage.
Syndrome polymalformatif, de transmission autosomique dominante à forte pénétrance et expressivité variable lié à des mutations du gène PTCH1.
- – Peau : apparition précoce de multiples carcinomes basocellulaires (siégeant dans 25 % des cas au niveau des paupières), kératose palmoplantaire, kystes des mâchoires.
- – Œil : colobome choriorétinien, cataracte, mélanocytomes rétiniens.
Vascularite d'étiologie inconnue atteignant les muqueuses, les yeux, la peau, les articulations et le système nerveux.
- – Peau : aphtes bipolaires, érythème noueux, pseudo-folliculite.
- – Œil : uvéite non granulomateuse synéchiante à hypopion, vascularites rétiniennes occlusives, papillite, œdème papillaire sur thrombophlébite cérébrale, paralysie oculomotrice.
Pathologie multisystémique de cause inconnue aboutissant à la formation de granulomes immunitaires. L'atteinte pulmonaire est prédominante.
- – Peau : expression clinique très variable dont érythème noueux, sarcoïdes cutanées.
- – Œil : uvéite antérieure granulomateuse, uvéite intermédiaire, périphlébite rétinienne, granulomes rétiniens et choroïdiens, choroïdite multifocale, œdème papillaire ou névrite optique.
Anomalie de synthèse du collagène ou de l'élastine touchant l'œil, la peau, le tissu squelettique et le système cardiovasculaire.
Six formes cliniques, dont la dernière nommée fragilitas oculi, comporte une atteinte oculaire grave et fréquente. L'hyperextensibilité articulaire et ligamentaire est un dénominateur commun.
- – Peau : peau fine, laxe mais fragile, cicatrices en papier à cigarette en regard des éminences osseuses.
- – Œil : sclérotiques bleutées, hyperlaxité palpébrale avec luxation et ptosis, épicanthus, fragilité oculaire globale pour le type VI (kératocône, troubles de la cicatrisation, subluxation cristallinienne, myopie forte avec staphylome, stries angioïdes, rupture oculaire pour des traumatismes minimes).
Transmission autosomique dominante. Anomalie de la fibrilline avec atteinte squelettique, vasculaire, ophtalmologique et cutanée.
- – Peau : vergetures horizontales dorsales, scapulaires et fessières.
- – Œil : sclérotiques bleutées, ectopie cristallinienne, myopie forte axile, décollement de rétine, kératocône, dégénérescence marginale pellucide.
Affection génétique rare touchant la peau, l'œil et le système vasculaire.
- – Peau : « peau de poulet plumé » avec papulo-nodules jaunâtres confluant en plaques réticulées au niveau du cou, des aisselles, de la région inguinale et des creux poplités.
- – Œil : sclérotiques bleutées, myopie forte axile, stries angioïdes multiples bilatérales avec risque majeur de néovascularisation vers la quatrième décennie.
Pathologie génétique liée à la mutation d'un gène suppresseur de tumeur (NF1). Présentation clinique très variable avec une atteinte cutanée, ophtalmologique, neurologique, squelettique, etc.
- – Peau : taches café au lait, neurofibromes cutanés ou sous-cutanés, lentigines des plis axillaires et inguinaux.
- – Œil : nodules de Lisch, gliome du nerf optique, névrome plexiforme de la paupière supérieure, glaucome, dysplasie orbitaire.
Pathologie génétique par mutation du gène TSC1 ou TSC2 aboutissant à la production d'hamartomes multisystémiques. Manifestations cutanées, neurologiques, rénales, pulmonaires, cardiaques et ophtalmologiques
- – Peau : angiofibromes en particulier des sillons nasogéniens, fibromes unguéaux (tumeurs de Koenen), taches achromiques en « feuille de sorbier » , plaques en peau de chagrin.
- – Œil : hamartomes rétiniens, tâches rétiniennes achromatiques.
Déficit enzymatique touchant le métabolisme de la méthionine. Transmission autosomique récessive. Atteinte squelettique (allure marfanoïde, scoliose, ostéoporose, etc.), neurologique (retard mental, épilepsie), cutanée et ophtalmologique.
- – Peau : faciès caractéristique avec cheveux fins, grisonnants, clairsemés, pommettes rouges.
- – Œil : ectopie cristallinienne inféronasale bilatérale asymétrique, microsphérophaquie avec risque d'ectopie et de glaucome aigu, myopie forte, décollement de rétine.
Pathologie de surcharge de transmission récessive liée à l'X nommée angiokératose diffuse universelle. Déficit enzymatique entraînant l'accumulation intralysosomiale de glycosphingolipides au sein des cellules de l'endothélium vasculaire. Débute tôt dans l'enfance : atteinte neurologique, rhumatologique, digestive, rénale, cardiaque, ORL.
- – Peau : angiokératomes en « caleçon » , acroparesthésies paroxytiques.
- – Œil : lésions constantes, y compris chez les femmes vectrices. Cornée verticillée avec opacités cornéennes disposées autour d'un axe inférieur, dilatations anévrismales des vaisseaux conjonctivaux, cataracte postérieure en rayons de roue, tortuosité des vaisseaux rétiniens, œdème palpébral, rarement rétinite pigmentaire et occlusions vasculaires.
Syndrome héréditaire de vieillissement précoce lié à la mutation du gène WRN (codant pour des protéines avec une activité hélicase), à transmission autosomique récessive.
- – Peau : scléropoïkilodermie avec atrophie, canitie précoce, alopécie progressive, faciès caractéristique avec nez en bec d'oiseau, raucité de la voix.
- – Œil : cataracte sous-capsulaire postérieure bilatérale, poliose, madarose, dégénérescence paramaculaire.
Pathologie rare, sporadique entraînant une atteinte cutanée, ophtalmologique, neurologique, squelettique, cardiovasculaire et urogénitale.
- – Peau : hamartome épidermique avec hyperplasie épidermique localisée, présente dès la naissance.
- – Œil : colobome, cataracte, microphtalmie, anophtalmie, dermolipomes conjonctivaux, choristomes, opacités cornéennes, troubles neurologiques avec cécité corticale.
Xanthogranulome juvénile :
- – peau : lésions papulonodulaires touchant le nourrisson;
- – œil : iris fréquemment atteint avec risque d'hyphéma spontané, glaucome par fermeture de l'angle.
- – BrÛlures thermiques ou chimiques atteignant les paupières : recherche systématique de brÛlure conjonctivocornéenne associée.
- – Herpès palpébral, zona V1 : recherche kérato-uvéite.
- – Syndrome de nécrolyse épidermique toxique (Lyell, Stevens-Jonhson) : évaluation et surveillance de l'atteinte ophtalmologique.
- – Kawasaki : urgence diagnostique.
- – Rétinoïdes : HTIC avec œdème papillaire, sécheresse, kératite, myopisation.
- – Anti-histaminiques : sécheresse, mydriase, crises oculogyres.
- – Rétinopathie aux antipaludéens de synthèse.
- – Corticoïdes locaux ou généraux : hypertonie oculaire, glaucome cortisonique, infection, cataracte.
- – Caroténoïdes (canthaxanthine) : rétinopathie en paillettes d'or.
N. André, M. Beylerian, M. -A. Heng, A. Aziz-alessi , D. Denis
La prise en charge des enfants présentant une pathologie hémato-oncopédiatrique est multidisciplinaire. L'ophtalmologiste fait partie des partenaires qui participent dans la prise en charge de ces patients.
Cette collaboration intervient dans les trois principaux temps du traitement :
- – le diagnostic initial;
- – le traitement;
- – la surveillance à moyen et long terme.
Bien que les tumeurs orbitaires soient rares en pédiatrie, elles peuvent être associées à une morbidité significative et à un risque de mortalité. Il peut s'agir de tumeurs bénignes ou malignes. Les tumeurs bénignes, telles que les kystes dermoïdes orbitaires, les lymphangiomes ou les hémangiomes capillaires, ne sont pas traités en hématologie et oncologie pédiatriques. Les pathologies malignes les plus fréquentes sont les rétinoblastomes, les rhabdomyosarcomes, les métastases et autres tumeurs rares.
Le rétinoblastome est la tumeur intra-oculaire la plus fréquente chez les enfants [1]. Au début du siècle passé, la survie d'un enfant atteint de rétinoblastome était exceptionnelle, aujourd'hui près de 95 % des cas de rétinoblastome peuvent être guéris [2]. Ce palier a pu être franchi grâce à la mise en place du dépistage précoce et de nouvelles stratégies thérapeutiques et grâce à une prise en charge pluridisciplinaire entre pédiatres, ophtalmologistes et oncologues, clé de voÛte du succès thérapeutique. Une fois le diagnostic de rétinoblastome posé, les enfants sont adressés à un centre de référence (en France, l'institut Curie à Paris) pour la réalisation d'un bilan d'évaluation et d'extension codifié à l'échelle nationale. Ce bilan doit être réalisé initialement pour pouvoir ensuite proposer la prise en charge la plus adaptée possible (cryothérapie couplée à la chimiothérapie, laser, injection intra-artérielle, énucléation, confections des prothèses, etc.) (voir chapitre 20).
Il existe également des pathologies plus rares primitives comme les rhabdomyosarcomes [3], les lymphomes, les tumeurs myofibroblastiques, les métastases de neuroblastomes ou de leucémies. Ainsi, si un enfant se plaint de symptômes ophtalmologiques (baisse de l'acuité visuelle, métamorphopsies, phosphènes, etc.), il devra bénéficier d'un examen ophtalmologique en urgence avec réalisation d'un fond œil pouvant se réaliser au bloc opératoire, selon son âge, afin de mettre en évidence la présence ou non de métastases intra-oculaires, d'infiltrats sous-rétiniens ou de hyalite signant une inflammation chronique.
Un bilan ophtalmologique est indispensable au moindre signe clinique, avec une attention particulière sur l'apparition d'une déviation oculomotrice (notamment si elle est accompagnée de céphalées, nausées et vomissements). L'examen ophtalmologique peut mettre en évidence une paralysie oculomotrice du nerf VI, ainsi qu'un œdème papillaire de stase bilatéral qui confirme le diagnostic d'HTIC. Il est important de rappeler que la normalité de l'examen clinique ophtalmologique n'élimine pas une hyperpression intracrânienne.
Les traitements anticancéreux peuvent entraîner des atteintes ophtalmologiques [4]. Elles peuvent être directes et/ou liées à l'immunodépression. Aussi, des complications ophtalmologiques peuvent être observées à type de rétinites virales, de conjonctivites – notamment sous cytarabine (Aracytine) à haute dose – et de maladie du greffon contre l'hôte post-allogreffe. Leur prise en charge requiert une collaboration étroite entre pédiatre et ophtalmopédiatre. L'apparition des nouvelles molécules thérapeutiques doit également nous rendre très vigilants sur leur impact potentiel sur la vision (par exemple inhibiteurs de Mitogen-activated Extracellular-signal-regulated Kinase [MEK] pouvant entraîner des décollements séreux rétiniens et nécessitant un suivi rapproché).
Par ailleurs, le suivi des symptômes initiaux doit être assuré rigoureusement. Ceci est particulièrement important pour les patients porteurs d'un gliome de bas grade envahissant les voies optiques pour lesquels le retentissement fonctionnel visuel prédomine sur le risque vital. L'évaluation radiologique n'est pas prédictive du devenir visuel, aussi une évaluation régulière est le facteur de surveillance le plus important [5].
L'augmentation des taux de survie en onco-hématologie pédiatrique impose une surveillance et vigilance accrue dans tous les domaines, notamment en ophtalmologie. De façon plus spécifique, le dépistage et la prise en charge de complications à court ou moyen terme comme le strabisme, les cataractes liées à l'utilisation de la chimiothérapie et l'irradiation sont recommandés par le Children Oncology Group [6].
Ne pas méconnaître les signes d’appel : strabisme, paralysie oculomotrice, diplopie, leucocorie, exophtalmie, baisse d’acuité visuelle inexpliquée.
Savoir évoquer en priorité :
-
tumeurs primitives : rétinoblastome, rhabdomyosarcome, lymphome, tumeur myofibroblastique ;
tumeurs secondaires : métastases de leucémies ou de neuroblastomes.
Prise en charge multidisciplinaire.
Importance du suivi pour les complications en cours de traitement et à distance.
[1] Gatta G, Rossi S, Aarelaid T, EUROCARE Working Group Childhood cancer survival in Europe 1999-2007 : results of EUROCARE-5–a population-based study Lancet Oncol: ( 2014 ) : 15: 35-47
[2] Rodriguez-Galindo C, Orbach DB, VanderVeen D Retinoblastoma Pediatr Clin North Am: ( 2015 ) : 62: 201-223
[3] Boutroux H, Levy C, Mosseri V Long-term evaluation of orbital rhabdomyosarcoma in children Clin Experiment Ophthalmol: ( 2015 ) : 43: 12-19
[4] Horwitz M, Auquier P, Barlogis V Incidence and risk factors for cataract after haematopoietic stem cell transplantation for childood leukaemia : an LEA study Br J Haematol: ( 2015 ) : 168: 518-525
[5] Dodgshun AJ, Elder JE, Hansford JR Long-term visual outcome after chemotherapy for optic pathway glioma in children : site and age are strongly predictive Cancer: ( 2015 ) : 121: 4190-4196
[6] Children's Oncology Group Long-term follow-up guidelines for survivors of childhood, adolescent, and young adult cancer. Version 4.0 – October 2013. En ligne : www.survivorshipguidelines.org/pdf/LTFUGuidelines_40.pdf
C. Oudin, M. Beylerian
Pédiatres et ophtalmologistes sont amenés à collaborer pour la prise en charge diagnostique et thérapeutique de certaines situations hématologiques.
Dans le domaine de l'hématologie maligne, certaines leucémies aiguës (myéloïdes ou lymphoïdes) de l'enfant peuvent se présenter, au diagnostic initial ou lors d'une rechute, avec des atteintes ophtalmologiques. Ces atteintes concernent le plus souvent les enfants porteurs de leucémies aiguës myéloïdes. Ces atteintes ophtalmologiques, symptomatiques ou non, se rencontrent à une fréquence variable selon les études publiées dans la littérature et concernent 15 à 20 % des enfants pour les séries pédiatriques les plus récentes [1, 2]. Il peut s'agir soit d'atteintes liées à l'infiltration par des cellules blastiques d'une structure oculaire, soit d'atteintes indirectes consécutives aux anomalies hématologiques secondaires à la leucémie aiguë.
L'infiltration par des cellules blastiques peut concerner quasiment toutes les structures ophtalmologiques (iris, choroïde, nerf optique, rétine) et orbitaires. Un œdème papillaire pourra être visible dans les situations d'infiltration du nerf optique. Dans les cas de localisation neuroméningée de la leucémie, il n'est pas rare d'observer des manifestations ophtalmologiques : diplopie par atteinte des nerfs oculomoteurs, œdème papillaire par HTIC. Le traitement est alors celui de la leucémie. Une atteinte oculaire isolée par infiltration blastique peut être le mode de révélation d'une rechute. Dans ce cas, la prise en charge thérapeutique comportera, outre la chimiothérapie, une irradiation de la cavité orbitaire afin de prévenir une nouvelle rechute localisée.
Parmi les atteintes consécutives aux anomalies hématologiques associées à la leucémie aiguë, les hémorragies rétiniennes (secondaires à une thrombopénie) sont les plus fréquemment rencontrées, même si l'on peut également observer d'autres types d'hémorragies : sous-conjonctivales, du vitré, etc. Des cas de ptosis secondaires à un hématome rétrobulbaire sur thrombopénie ont été rapportés [1]. Ont également été décrits de rares phénomènes d'occlusion vasculaire artérielle ou veineuse (occlusion de l'artère ou de la veine centrale de la rétine), secondaires à la leucostase et à l'hyperviscosité sanguine qui en découle, rencontrée dans certaines leucémies aiguës très hyperleucocytaires. Cette atteinte est également classique au diagnostic de leucémie myéloïde chronique (pathologie exceptionnelle chez l'enfant).
Au total, il n'est à ce jour pas recommandé en France de réaliser un examen ophtalmologique de manière systématique dans les cas de nouveau diagnostic de leucémie aiguë de l'enfant ou de l'adulte. Le recours à l'expertise ophtalmologique est en revanche indispensable devant tout point d'appel clinique évocateur.
Les traitements des leucémies aiguës peuvent se compliquer d'atteintes ophtalmologiques. La cataracte est en effet extrêmement fréquente après greffe de cellules souches hématopoïétiques, particulièrement dans les cas de préparation comportant une irradiation corporelle totale [3], ce qui implique une surveillance ophtalmologique rigoureuse dans ce type de situation. Ce risque et les conséquences en termes de dépistage concernent aussi les cas devenus rares dans lesquels le traitement de la leucémie comporte une irradiation du système nerveux central. Il semblerait en revanche que l'usage de la corticothérapie dans le cadre du traitement des leucémies impacte peu la survenue de cataracte [4].
Le glaucome est également une complication grave du traitement par corticothérapie qui peut passer inaperçue du fait de l'absence de symptomatologie rapportée par l'enfant. La fréquence est variable selon les études [5, 6], le niveau pressionnel peut aller de plus de 21 mmHg à des chiffres très élevés (42 mmHg). La technique de mesure de la tension oculaire est importante à préciser, car la tonométrie non-contact peut surestimer les valeurs chez l'enfant par manque de coopération : il est donc indispensable d'effectuer cette prise de tension à aplanation même si la coopération de l'enfant là encore peut être parfois difficile [6]. La possibilité d'une hypertension silencieuse et ses conséquences irréversibles d'atrophie optique, et donc de cécité, doit être connue par l'oncopédiatre qui doit pouvoir rechercher à l'interrogatoire les signes fonctionnels d'inconfort oculaire (douleurs, vision floue, conjonctivite, etc.) et de céphalées.
Un autre type de complication ophtalmologique classique de la transplantation de cellules souches hématopoïétiques correspond à la survenue d'une maladie du greffon contre l'hôte de localisation oculaire. Celle-ci se traduit classiquement par un syndrome sec oculaire, parfois très invalidant associé à des conjonctivites pseudo-membraneuses. Le traitement consiste en des collyres lubrifiants et des agents mouillants; si nécessaire dans la gradation thérapeutique, on peut utiliser des collyres immunosuppresseurs (ciclosporine 0,1 % : Ikervis, ou plutôt en préparation hospitalière à 1 ou 2 % ); un collyre au sérum autologue en préparation hospitalière a un effet trophique sur la surface oculaire. Ponctuellement, on peut avoir recours à une corticothérapie locale. En cas de présence de pseudo-membranes, il sera indispensable de les retirer à la pince, pour limiter la noncicatrisation d'un ulcère de cornée associé et pour éviter, plus tard, la survenue d'une fibrose tarsale conjonctivale (voir chapitre 8). Des verres scléraux sont parfois utiles mais difficiles d'utilisation.
À la découverte d'un purpura thrombocytopénique auto-immun, cause la plus fréquente de thrombopénie profonde isolée en pédiatrie, le taux de plaquettes est souvent extrêmement abaissé, inférieur à 20 G/L dans plus de deux tiers des cas. Or, le risque d'hémorragie sévère est corrélé à la profondeur de la thrombopénie. Le fond d'œil, à la recherche d'hémorragies intrarétiniennes et surtout d'un œdème papillaire évocateur d'une HTIC sur hémorragie intracrânienne, peut donc parfois être utile, même s'il n'est pas recommandé de manière systématique [7].
Concernant les maladies hémorragiques héréditaires (hémophilie, maladie de Willebrand), les manifestations hémorragiques oculaires en pédiatrie sont rares, même si quelques cas spontanés d'hémorragies rétrobulbaires et de saignements de la chambre antérieure de l'œil chez des hémophiles ont été décrits. De même, en pédiatrie, les pathologies ophtalmologiques consécutives à une prédisposition héréditaire aux accidents thrombotiques (thrombophilie) sont exceptionnelles. On peut néanmoins décrire, du fait de sa gravité, le déficit homozygote en protéine C qui peut se manifester de façon rarissime par l'existence de thromboses (anténatales) des vaisseaux rétiniens [8]. La cécité est une séquelle majeure chez ces enfants.
La rétinopathie du patient porteur d'un syndrome drépanocytaire majeur, qu'elle soit proliférante ou non, est une complication classique de cette hémoglobinopathie, particulièrement chez les patients porteurs d'une drépanocytose SC. En France, un suivi ophtalmologique annuel est donc recommandé dès l'âge de 6 ans pour les patients porteurs de drépanocytose SC et de 10 ans pour ceux porteurs d'une drépanocytose SS [9]. En effet, il est classique d'opposer, au sein des syndromes drépanocytaires majeurs, la forme homozygote SS des formes doubles hétérozygotes SC dont la maladie systémique est moins sévère et moins invalidante, mais dont l'atteinte rétinienne est plus fréquente.
Les manifestations ophtalmologiques de la drépanocytose sont :
- – au niveau de la conjonctive : vaso-occlusion du réseau conjonctival bulbaire inférieur (aspect des vaisseaux conjonctivaux en « tire-bouchon » );
- – au niveau de la chambre antérieure et de l'iris : hyphémas traumatiques, avec risque d'hypertonie plus élevé que chez les sujets indemnes; un lavage chirurgical précoce de la chambre antérieure est souvent nécessaire;
- – au niveau rétinien : rétinopathie drépanocytaire typique par occlusion des capillaires rétiniens périphériques liée à la falciformation des hématies. Il en résulte une capillaropathie ischémique, qui affecte généralement la périphérie temporale. D'abord transitoires, ces occlusions entraînent une souffrance pariétale, à l'origine d'hémorragies rétiniennes, puis une ischémie chronique avec développement d'anastomoses artérioveineuses puis de néovascularisation par libération de facteurs angiogéniques. Elle est classée en :
-
rétinopathie non proliférante (oedème ischémique intrarétinien, « hémorragies saumonées » , microdépôts jaunâtres brillants appelés « givre doré » qui sont des macrophages chargés d’hémosidérine) ; >
rétinopathie proliférante (stade I : occlusions artériolaires périphériques ; stade II : anastomoses artérioveineuses ; stade III : néovascularisation rétinienne périphérique (sea fan) ; stade IV : hémorragie intravitréenne ; stade V : décollement de rétine).
La littérature est très abondante concernant les atteintes infectieuses ophtalmologiques à germe opportuniste chez les patients infectés par le VIH porteurs d'un syndrome d'immunodéficience acquise. Ces complications peuvent également survenir dans certains déficits immunitaires primitifs. On peut citer pour exemple la toxoplasmose oculaire, la rétinopathie à CMV ou encore les infections fongiques oculaires (aspergillose, etc.) : ce type d'atteinte est néanmoins peu fréquent. Les cas d'herpès et de zona ophtalmiques ne sont pas exceptionnels chez les patients porteurs d'un déficit immunitaire primitif humoral ou acquis suite à une greffe de cellules souches hématopoïétiques ou au traitement d'une leucémie aiguë.
Hémorragie rétinienne et thrombopénie.
Cataracte et irradiation dans le cadre de la prise en charge des leucémies aiguës.
Glaucome et corticothérapie.
Rétinopathie et drépanocytose.
[1] Bitirgen G, Belviranli S, Caliskan U Ophthalmic manifestations in recently diagnosed childhood leukemia Eur J Ophthalmol: ( 2016 ) : 26: 88-91
[2] Reddy SC, Jackson N, Menon BS Ocular involvement in leukemia–a study of 288 cases Ophthalmologica: ( 2003 ) : 217: 441-445
[3] Horwitz M, Auquier P, Barlogis V Incidence and risk factors for cataract after haematopoietic stem cell transplantation for childhood leukaemia : an LEA study Br J Haematol: ( 2015 ) : 168: 518-525
[4] Alloin AL, Barlogis V, Auquier P Prevalence and risk factors of cataract after chemotherapy with or without central nervous system irradiation for childhood acute lymphoblastic leukaemia : an LEA study Br J Haematol: ( 2014 ) : 164: 94-100
[5] Yamashita T, Kodama Y, Tanaka M Steroid-induced glaucoma in children with acute lymphoblastic leukemia : a possible complication J Glaucoma: ( 2010 ) : 19: 188-190
[6] de Queiroz Mendonca C, de Souza CP, Martins-Filho PR Steroid-induced ocular hypertensive response in children and adolescents with acute lymphoblastic leukemia and non-Hodgkin lymphoma Pediatr Blood Cancer: ( 2014 ) : 61: 2083-2085
[7] Société française d'hématologie pédiatrique Purpura thrombopénique idiopathique Archives de Pédiatrie: ( 2007 ) : 14: 1394-1398
[8] Dreyfus M, Ladouzi A, Chambost H Treatment of inherited protein C deficiency by replacement therapy with the French purified plasma-derived protein C concentrate (PROTEXEL) Vox Sang: ( 2007 ) : 93: 233-240
[9] Haute Autorité de santé (HAS) ; janvier 2010. En ligne : http://www.has-sante.fr/portail/upload/docs/application/pdf/2010-04/ald_10_pnds_drepano_enfant_web.pdf
E. Marquand, R. Reynaud, E. Bui Quoc
Les pédiatres spécialisés en endocrinologie font appel aux ophtalmologistes principalement dans certaines situations cliniques telles que le diabète, l'obésité, les retards de croissance staturale syndromiques ou d'autres pathologies plus rares (fig. 27-1 et tableau 27-1).
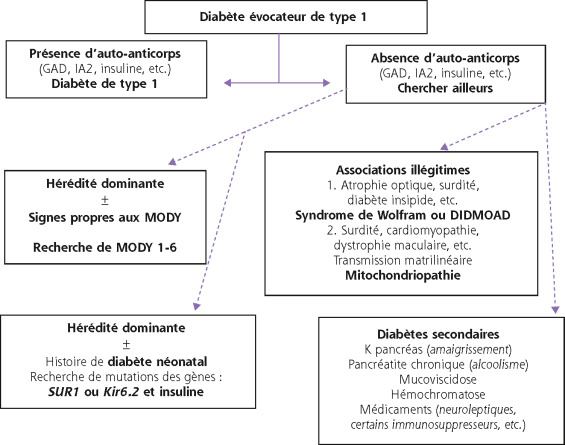
fig. 27-1 Manifestations ophtalmologiques à rechercher dans certaines pathologies endocriniennes.
DIDMOAD : diabetes insipidus, diabetes mellitus, optic atrophy, and deafness ; GAD : glutamate acide décarboxylase ; IA : islet antigen 2 ; MODY : maturityonset diabetes of the young.
Tableau 27-1 – Pathologies endocriniennes et signes ophtalmologiques associés.
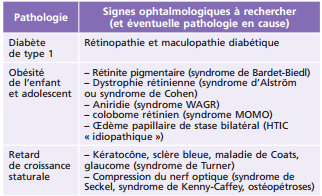
HTIC : hypertension intracrânienne ; MOMO : macrosomie foetale, obésité, macrocéphalie et anomalies oculaires ; WAGR : Wilms tumor, aniridia, genital anomalies, mental retardation.
Le diabète de type 1, deuxième maladie chronique de l'enfant après l'asthme, connaît une augmentation mondiale d'incidence allant de 3 à 5,5 % par an depuis le début du 3 millénaire (en France, + 5,25 % par an sur 4 ans à la fin des années 2000). Ainsi, 1 800 enfants de moins de 15 ans deviennent diabétiques en France chaque année. Cette augmentation d'incidence est plus importante dans le groupe d'âge le plus jeune (0-4 ans); elle est plus modérée dans le groupe d'âge des adolescents (10-14 ans). Ainsi le nombre d'enfants de moins de 5 ans devenant diabétiques a doublé en 30 ans dans de nombreux pays et cette augmentation devrait continuer dans les prochaines décennies. Au total, le nombre de nouveaux cas passera en Europe de 15 000 en 2005 à 25 000 en 2020 et le nombre total d'enfants de moins de 15 ans ayant un diabète passera de 94 000 en 2005 à 160 000 en 20202.
Dans le cadre de la prise en charge du diabète de l'enfant, l'ophtalmologiste est sollicité :
- – lors du bilan initial du diabète, pour établir un état des lieux d'éventuelles anomalies rétiniennes préexistantes associées ou mettre en évidence une rétinopathie diabétique éventuellement déjà présente. En cas de suspicion de diabète non auto-immun – absence d'auto-anticorps GAD (glutamate acide décarboxylase), IA2 (islet antigen 2), insuline –, l'examen ophtalmologique peut retrouver des anomalies orientant vers un diagnostic (par exemple : atrophie optique dans le syndrome de Wolfram, dystrophie maculaire dans les mitochondriopathies)3 (fig. 27-1);
- – lors du suivi, pour dépister la rétinopathie diabétique et/ou la maculopathie diabétique, première cause de cécité acquise de l'adulte avant 50 ans. Le dépistage des formes débutantes est primordial car cette complication reste longtemps asymptomatique. La rétinopathie diabétique en France peut survenir à partir de 5 à 10 ans d'évolution du diabète [1] mais la période pubertaire reste une période à haut risque évolutif. Aussi, si la rétinopathie proliférante est rare avant 20 ans, certaines études rapportent entre 34 et 42 % de prévalence de la rétinopathie diabétique en période pubertaire [2]. Les recommandations actuelles sont de réaliser un examen ophtalmologique avec premier examen du fond de l'œil à partir de l'âge de 12 ans et examen obligatoirement annuel à partie de l'âge de 15 ans. La vigilance est accrue en période pubertaire. En cas de déséquilibre métabolique majeur avec intensification de la prise en charge, cette surveillance est rapprochée de manière trimestrielle ou semestrielle en période pubertaire [3]. La réalisation de rétinophotographies est plus sensible que l'examen clinique (voir chapitre 29.10).
Inversement dans le cadre de la prise en charge ophtalmologique, en présence d'une rétinopathie débutante ou rapidement évolutive, l'ophtalmologiste peut demander une modification du traitement du diabète.
1. Source Éco-santé régions et départements 2010, régime général.
2. Source Eurodiab.
L'obésité de l'enfant et de l'adolescent n'échappe pas à la pandémie décrite pour la population adulte. Selon les données de l'Organisation mondiale de la santé (OMS), 43 millions d'enfants et adolescents sont en surpoids et obésité dans le monde en 2016n avec une estimation de prévalence de 9,1 % en 2020 soit 60 millions d'enfants. En France, la prévalence est passée de 5 % dans les années 1980 à 16 % au début des années 2000.
Les différents plans nationaux (Plan national nutrition santé puis Plan national obésité) ont fait stopper cette ascension sans amorcer une décroissance. Le dernier rapport de la Direction de la recherche, des études, de l'évaluation et des statistiques (DREES) 2015 rapporte une prévalence en France du surpoids et de l'obésité de 12,2 % pour les grandes sections de maternelle (« 8,7 % des élèves scolarisés en grande section de maternelle (5-6 ans) en 2012-2013 sont en surpoids et 3,5 % sont obèses » ), de 18,8 % pour les CM2 et de 17,6 % pour les élèves en 3 (« respectivement 14,8 % et 13,7 % sont en surpoids et […] l'obésité concerne 4,0 % et 3,9 % des élèves » ).
Lors de la prise en charge initiale, il est essentiel de différencier les obésités dites « communes » , liées au mode de vie, des obésités d'origine génétique, monogéniques ou syndromiques. Ainsi, en présence d'une obésité infantile ou associée à des troubles de développement, le pédiatre demandera un examen ophtalmologique avec examen du fond de l'œil et électrorétinogramme à la recherche de certaines anomalies caractéristiques des syndromes suivants :
- – rétinite pigmentaire dans le syndrome de Bardet-Biedl (1/125 000 à 1/175 000 patients) ou dans le syndrome d'Alstrom; – dystrophie rétinienne dans le syndrome de Cohen;
- – aniridie dans le syndrome WAGR (Wilms tumor, aniridia, genital anomalies, mental retardation) : < 1/100 000;
- – colobome rétinien dans le rare syndrome « MOMO » : macrosomie fœtale (surpoids à la naissance), obésité, macrocéphalie (hypertrophie de la tête) et anomalies oculaires (colobome et nystagmus) [4].
Par ailleurs, certaines complications de l'obésité sévère requièrent l'intervention des ophtalmologistes, telles que l'HTIC idiopathique objectivée sur le plan ophtalmologique par un œdème papillaire de stase bilatéral. Cette complication déjà connue dans les populations d'obèses adultes commence à être rapportée chez des adolescents et nécessite une prise en charge active et spécialisée [5]. L'examen du fond d'œil, l'analyse en optical coherence tomography (OCT) retinal nerve fiber layer (RNFL) et le champ visuel sont alors essentiels pour caractériser l'importance de l'œdème papillaire et assurer le suivi évolutif au cours de la prise en charge.
Les retards de croissance staturale constituent une cause fréquente de consultation. Lorsqu'une cause endocrinienne ou osseuse est évoquée, l'examen ophtalmologique est requis dans diverses situations cliniques, initialement et au cours du suivi :
- – pour les patientes avec syndrome de Turner (prévalence chez les filles : 1/2500 naissances) : les anomalies oculaires touchent plus de 50 % des jeunes filles porteuses de syndrome de Turner [6] et doivent donc être systématiquement recherchées. On peut retrouver un kératocône, une sclère bleue et/ou une maladie de Coats;
- – pour les patients atteints de pathologie hypophysaire :
en cas de tumeur hypophysaire, les conséquences fonctionnelles visuelles doivent être recherchées (compression du chiasma) ;
en cas de pathologie hypophysaire syndromique avec déficit hormonal : en effet, 16 % des patients avec déficit hypophysaire multiple constitutionnel ont une malformation oculaire ou oculomotrice associée [7].
- -pour les patients avec pathologie osseuse telle que le syndrome de Seckel (forme de nanisme ostéodysplasique avec microcéphalie), le syndrome de Kenny-Caffey (pathologie squelettique avec en particulier épaississement des corticales osseuses) ou les ostéopétroses, un examen ophtalmologique est requis pour rechercher en particulier des signes de compression du nerf optique.
Parfois, c'est l'ophtalmologiste qui sollicite l'endocrinologue pédiatre : en effet, c'est lui qui va être confronté par exemple à une perte de champ visuel pouvant faire découvrir une tumeur hypophysaire. Un bilan pédiatrique est également requis dans le cadre de malformations congénitales ou infantiles du globe oculaire (cataracte, strabisme, microphtalmie, dystrophie rétinienne, colobome) ou des nerfs optiques (hypoplasie ou aplasie), pouvant être liées à une pathologie endocrinienne (par exemple dysplasie septo-optique, syndrome CHARGE [Coloboma, Heart defect, Atresia choanae, Retarded growth, Genital anomalies, Ear anomalies], morning glory syndrome avec anomalies associées de la ligne médiane, etc.).
D'autres pathologies doivent favoriser un dialogue étroit entre ophtalmologiste et endocrinologue pédiatre tel le syndrome triple A (maladie multisystémique très rare caractérisée par une insuffisance surrénale avec un déficit isolé en glucocorticoïdes, une achalasie, une alacrymie, une dysfonction autonomique et une neurodégénerescence) afin de dépister une insuffisance surrénalienne latente devant une alacrymie.
Il est important de caractériser au mieux le phénotype clinique de ces patients au sein d'une équipe multidisciplinaire afin de dépister les déficits latents, coordonner les prises en charge et orienter les recherches génétiques.
Remerciements : Marie Beylerian, Valentine Bautrant.
[1] Donaghue KC, Fung AT, Hing S The effect of prepubertal diabetes duration on diabetes microvascularcomplications in early and late adolescence Diabetes Care: ( 1997 ) : 20: 77-80
[2] Klein R, Klein BE, Moss SE Retinopathy in young-onset diabetic patients Diabetes Care: ( 1985 ) : 8: 311-315
[3] Donaghue KC, Wadwa RP, Dimeglio LA ISPAD Clinical Practice Consensus Guidelines 2014. Microvascular and macrovascular complications in children and adolescents Pediatric Diabetes: ( 2014 ) : 15: 257-269
[4] Mason K, Page L, Balikcioglu PG Screening for hormonal, monogenic and syndromic disorders in obese infants and children Pediatr Ann: ( 2014 ) : 43: 218-224
[5] Paley GL, Sheldon CA, Burrows EK Overweight and obesity in pediatric secondary pseudotumor cerebri syndrome Am J Ophthalmol: ( 2015 ) : 159: 344-352
[6] Wikiera B, Mulak M, Koltowska-Haggstrom M The presence of eye defects in patients with Turner syndrome is irrespective of their karyotype Clin Endocrinol (Oxf): ( 2015 ) : 83: 842-848
[7] Macchiaroli A, Kelberman D, Auriemma RS A novel heterozygous SOX2 mutation causing congenital bilateral anophthalmia, hypogonadotropic hypogonadism and growth hormone deficiency Gene: ( 2014 ) : 534: 282-285
A. Fabre, M. Beylerian
La collaboration entre les ophtalmologistes et les hépato-gastro-entérologues pédiatres peut prendre plusieurs formes, celle de l'aide au diagnostic du fait d'une atteinte ophtalmologique pathognomonique d'un syndrome, celle du suivi de maladies ayant une atteinte ophtalmologique associée. Ainsi, on peut citer deux exemples de maladies rares où l'examen ophtalmologique peut permettre leur diagnostic rapide : la maladie de Wilson et le syndrome d'Alagille [1].
La maladie de Wilson est une thésaurismose due à une accumulation toxique de cuivre tissulaire par mutation du gène ATP7B de transmission récessive [2]. La prévalence de la pathologie est de 1/30 000 à 1/100 000. Les conséquences principales sont une atteinte hépatique de début précoce pouvant entraîner une cirrhose et/ou une insuffisance hépatocellulaire aiguë nécessitant dans les cas les plus sévères une greffe hépatique. L'autre atteinte est une atteinte neurologique ou neuropsychiatrique généralement plus tardive pouvant débuter à l'adolescence et pouvant entraîner des manifestations neurologiques variées ou psychiatriques trompeuses. La présence de l'anneau de Kayser-Fleischer dans la maladie de Wilson est pathognomonique. Il est inconstant dans les formes hépatiques et, en revanche, constamment présent dans les formes neurologiques. Cet anneau de coloration grise ou dorée est la conséquence des dépôts de cuivre en périphérie de la cornée : il est visualisé grâce à l'examen à la lampe à fente. Cet examen simple et rapide permet de faire le diagnostic avant l'obtention des résultats biologiques et de commencer le traitement chélateur sans délai.
Le syndrome d'Alagille, maladie génétique autosomique dominante caractérisée par des mutations des gènes JAG1 ou NOTCH2, dont la prévalence est estimée à 1/70 000 [3], est caractérisé par la présence d'une paucité ductulaire hépatique (absence partielle ou totale des canaux biliaires interlobulaires) associée à au moins trois des cinq caractéristiques cliniques suivantes :
- – un embryotoxon postérieur (proéminence de la ligne de Schwalbe à l'examen à la lampe à fente; il est présent chez 78 à 89 % des personnes atteintes de syndrome d'Alagille);
- – une cholestase;
- – une dysmorphie faciale (front proéminent, yeux enfoncés avec hypertélorisme modéré, menton pointu et nez droit en forme de pointe);
- – une anomalie congénitale cardiaque dont la manifestation la plus fréquente est une sténose des artères pulmonaires;
- – des anomalies vertébrales (vertèbres en forme de papillon).
Des anomalies rénales et/ou vasculaires peuvent aussi se manifester.
D'autres manifestations ophtalmologiques peuvent apparaître dans le syndrome d'Alagille, comme une anomalie d'Axenfeld-Rieger, qui se caractérise par un embryotoxon associé à des ponts iriens et des corectopies ou polycories avec atrophie irienne en secteur.
La réalisation d'une biopsie hépatique n'est plus indispensable pour poser le diagnostic, si la cholestase est associée à au moins trois des manifestations suivantes : oculaires, cardiaques, faciales et/ou vertébrales. Cependant ce diagnostic est généralement confirmé par biologie moléculaire. Du fait des multiples atteintes, une prise en charge multidisciplinaire est indispensable pour poser le diagnostic.
Les exemples d'associations retrouvés sont la galactosémie et l'avitaminose A.
Il s'agit d'une maladie génétique autosomique récessive (mutation du gène GALT, prévalence entre 1/40 000 à 1/60 000 en France) suspectée chez un enfant présentant une insuffisance hépatocellulaire fréquemment dans un contexte de mauvaise croissance staturopondérale associée à des difficultés alimentaires et à un sepsis à point de départ urinaire à E. coli après l'introduction du lactose [4]. La présence d'une cataracte fait suspecter le diagnostic de galactosémie. En effet, 75 % des patients atteints de galactosémie vont développer une cataracte bilatérale, généralement au cours des premières semaines de vie. Le principal facteur initiant ce type de cataracte est la forte concentration de galactose dans l'humeur aqueuse : l'accumulation de galactose et de galactitol dans les cellules cristalliniennes conduit à une augmentation de la pression osmotique intracellulaire et un afflux de fluide dans le cristallin, ce qui opacifie la capsule postérieure et le noyau, donnant un aspect en « goutte d'huile » en rétro-illumination.
Le traitement de galactosémie comprend l'élimination du galactose de l'alimentation (notamment le lait et ses dérivés, le jaune d'œuf, les abats et certains fruits et légumes). Si la maladie reste non traitée, les cataractes progressent vers une opacification totale du cristallin. Si la maladie est diagnostiquée et traitée précocement, l'installation de la cataracte précoce peut être inversée grâce à une intervention diététique rigoureusement suivie.
De manière non spécifique, le déficit en vitamine A résultant des cholestases non substituées peut induire des héméralopies, des sécheresses conjonctivales et cornéennes qui peuvent se compliquer de taches de Bitôt (taches conjonctivales blanches ou jaunâtres de taille et forme variables, correspondant à une accumulation de débris de kératine situés superficiellement dans la conjonctive) et/ou d'ulcères de cornée (voir chapitre 9). Plus rarement, sont décrits des cas de compression du nerf optique par hyperostose.
Les associations ophtalmologiques les plus fréquentes sont celles liées aux maladies inflammatoires de l'intestin (maladie de Crohn et rectocolite hémorragique) et à leur traitement [4, 5].
Le diagnostic de ces maladies est clinique, endoscopique et histologique. Le traitement repose sur les dérivés salicylés, la corticothérapie, les immunosupresseurs et les anti-TNF. Dans ces maladies, l'atteinte ophtalmologique peut toucher jusqu'à 10 % des patients (plus fréquemment les filles), aussi bien en cas d'atteinte colique et iléocolique que d'atteinte isolée de l'intestin grêle.
Ces maladies inflammatoires intestinales peuvent atteindre tous les tissus oculaires; l'épisclérite aiguë et l'uvéite antérieure non granulomateuse sont les atteintes les plus fréquentes. Plus rarement, on retrouve une sclérite antérieure voire postérieure, des kératites, des conjonctivites, des neuropathies optiques ischémiques et des pseudo-tumeurs inflammatoires de l'orbite. Un suivi régulier et une attention accrue sont nécessaires.
La corticothérapie générale [6] est un traitement des crises des maladies inflammatoires de l'intestin; son utilisation doit être parcimonieuse du fait du risque sur la croissance et des effets indésirables.
Parmi ces derniers, il existe des complications ophtalmologiques spécifiques qui sont les suivantes :
- – hypertonie et/ou glaucome secondaire expliquant la nécessité de la mesure de la pression intra-oculaire (PIO) systématique chez tout patient sous cortisone (au moins 1 fois/an si corticothérapie générale et 1 fois tous les 3 mois si corticothérapie en collyre). L'arrêt de la corticothérapie locale peut suffire à faire descendre la PIO. L'hypertonie oculaire cortisonique peut évoluer vers un véritable glaucome secondaire;
- – cataracte sous-capsulaire postérieure, liée à la dose (souvent forte) et à la durée du traitement (souvent plusieurs années);
- – retard de cicatrisation cornéenne allant jusqu'au risque de perforation [7].
Les ophtalmologistes comme les gastro-entérologues doivent être systématiques dans leur démarche clinique et travailler en interdisciplinarité.
Le syndrome d’Alagille et la maladie de Wilson sont deux pathologies où l’interdisciplinarité est capitale entre hépato-gastro-entérologues et ophtalmopédiatres afin de poser rapidement un diag nostic.
Le suivi de patients ayant un risque d’atteinte ophtalmologique, du fait de l’histoire naturelle de la maladie ou du traitement, nécessite également une collaboration par la connaissance des risques et des moyens à mettre en oeuvre.
[1] O’Neill DP. The eye and liver disorders. Eye 1992 ; 6 : 366-70.
[2] Spinner NB, Leonard LD, Krantz ID. Alagille Syndrome. In : GeneReviews® [Internet]. Pagon RA, Adam MP, Ardinger HH, et al. Eds. Seattle (WA) : University of Washington, Seattle 1993–2016. 2000 May 19 [updated 2013 Feb 28].
[3] Weiss KH. Wilson Disease. In : GeneReviews® [Internet]. Pagon RA, Adam MP, Ardinger HH, et al. Eds. Seattle (WA) : University of Washington, Seattle 1993–2016. 1999 Oct 22 [updated 2016 Jul 29].
[4] Berry GT. Classic Galactosemia and Clinical Variant Galactosemia. In : Gene- Reviews® [Internet]. Pagon RA, Adam MP, Ardinger HH, et al. Eds. Seattle (WA) : University of Washington, Seattle 1993–2016. 2000 Feb 4 [updated 2014 Apr 3].
[5] Aloi M, Cucchiara S. Extradigestive manifestations of IBD in pediatrics. Eur Rev Med Pharmacol Sci 2009 ; 13 Suppl 1 : 23-32.
[6] Hofl ey P, Roarty J, McGinnity G, et al. Asymptomatic uveitis in children with chronic infl ammatory bowel diseases. J Pediatr Gastroenterol Nutr 1993 ; 17 : 397-400.
[7] Tripathi RC, Kipp MA, Tripathi BJ, et al. Ocular toxicity of prednisone in pediatric patients with infl ammatory bowel disease. Lens Eye Toxic Res 1992 ; 9 : 469-82.
C. Ovaert, A. Aziz-Alessi , D. Denis
En cardiopédiatrie, de nombreuses pathologies peuvent s'accompagner d'atteintes ophtalmologiques; celles-ci peuvent constituer le premier signe d'appel et aider au diagnostic étiologique ou, inversement, être recherchées dans le cadre d'un bilan diagnostique d'une affection cardiovasculaire.
Les malformations cardiaques s'inscrivent souvent dans des syndromes d'origine génétique affectant plusieurs organes. La cardiopathie peut être le point d'entrée. Le cardiologue devra alors, devant la suspicion d'une pathologie multisystémique, faire appel aux autres spécialistes dont les ophtalmologistes.
La liste suivante détaille les entités les plus fréquentes mais ne se veut pas exhaustive.
- - Syndrome de Marfan [1] : maladie génétique avec anomalie du tissu conjonctif. Le diagnostic est évoqué en présence d'une dilatation de la racine aortique ou d'une laxité anormale de la valve mitrale avec insuffisance. La mise en évidence par l'ophtalmologue d'une ectopie ou luxation du cristallin permet de conforter le diagnostic de syndrome de Marfan avant les résultats génétiques et de mettre en place un traitement par β-bloquants.
- – Syndrome CHARGE [2] : le terme CHARGE est un acronyme des critères majeurs de cette maladie = C : colobome (typiquement bilatéral choriorétinien et du nerf optique), H : anomalies cardiaques, A : atrésie/sténose choanale, R : retard croissance et développement, G : hypoplasie génitale, E : anomalies des oreilles et/ou surdité. Dans ce syndrome, les anomalies cardiaques, très fréquentes, peuvent être le point d'entrée car elles peuvent être sévères (large canal artériel, anomalies cono-troncales).
- – Syndrome d'Alagille [3] : en cas de sténose des artères pulmonaires ou de tétralogie de Fallot associée à une cholestase, la mise en évidence d'un embryotoxon conforte l'hypothèse d'un syndrome d'Alagille.
- – Microdélétion 22q11 [4] : le signe d'appel initial est principalement cardiaque car l'atteinte est souvent sévère de type conotroncale. L'évaluation ophtalmologique est indispensable pour un bilan complet, car les signes oculaires sont nombreux et très fréquents à type d'anomalie des paupières (télécanthus, hypertélorisme), sourcils et cils clairsemés, blépharite, dystrichiasis, embryotoxon postérieur, reliquats embryonnaires pupillaires, tortuosité vasculaire, anomalie réfractive, strabisme. Une cécité corticale peut également être rencontrée.
- – Trisomie 21 : il existe un certain nombre de malformations cardiaques et ophtalmologiques mais aucune n'est pathognomonique de la trisomie 21. Sur le plan cardiologique, par ordre de fréquence on citera le canal atrioventriculaire (CAV), la communication interventriculaire (CIV), la communication interauriculaire (CIA), la tétralogie de Fallot et la persistance du canal artériel. Ces malformations doivent être recherchées le cas échéant en prénatal, et bien sÛr après la naissance, par une échographie et devront être prise en charge médicalement et/ou chirurgicalement en milieu spécialisé en fonction de leur gravité. Sur le plan ophtalmologique, les signes oculaires sont nombreux et fréquents; ils vont de simples troubles de réfraction à des cataractes congénitales.
- – Syndrome de Williams-Beuren [5] : la sténose supravalvulaire aortique peut se présenter de façon isolée ou dans un contexte de syndrome de Williams-Beuren; elle entraîne un risque de bas débit coronaire en particulier en cas d'anesthésie générale qui doit être extrêmement précautionneuse. La mise en évidence d'anomalies ophtalmiques, de type iris stellaire (50 % des patients) mieux visible en cas d'iris clair, strabisme et/ou troubles de la réfraction, et d'anomalies vasculaires rétiniennes peut orienter vers le syndrome de Williams.
- – Les syndromes suivants sont plus rares et peuvent également associer des anomalies cardiaques et ophtalmologiques :
-
syndrome de Smith-Lemli-Opitz (cataracte, strabisme et nystagmus ; CIV, CIA, CAV, persistance du canal artériel) ;
neurofibromatose de type 1 (gliome du nerf optique, nodules de Lisch, neurofibrome plexiforme ; hypertension artérielle par atteinte rénale) ;
syndrome de Rubinstein-Taybi (obstruction des canaux lacrymonasaux, glaucome congénital, anomalies réfractives ; susceptibilité à l’anesthésie avec risque d’aryrhmie cardiaque) ;
syndrome de Waardenburg (télécanthus, anomalie pigmentaire de l’iris ; anomalies cardiovasculaires) ;
syndrome de Kabuki (sclérotique bleue, strabisme, ptose, colobome et anomalies de la cornée, fentes palpébrales allongées avec éversion de la partie latérale du 1/3 inférieur de la paupière ; lésions obstructives du coeur gauche ou CIV) ;
syndrome de Smith-Magenis (fentes palpébrales obliques en haut et en dehors dans plus de 60 % des cas, myopie, anomalies iriennes et rarement décollement rétinien ; malformations cardiaques).
Certaines malformations cardiaques peuvent être le résultat de la prise de toxiques. Citons en particulier le syndrome d'alcoolisme fœtal [6] qui peut associer une anomalie cardiaque potentiellement sévère (CIV, maladies conotroncales) et des signes ophtalmologiques : fente palpébrale étroite, ensellure nasale marquée, tortuosité vasculaire, colobome et microphtalmie.
Certaines atteintes cardiaques peuvent s'accompagner de complications oculaires :
- – l'endocardite infectieuse peut générer des embols ou des lésions de vasculite (purpura, taches de Roth);
- – les cardiomyopathies peuvent s'accompagner de thrombus intracavitaires (par exemple ventricule gauche non compacté) qui peuvent emboliser vers les vaisseaux rétiniens;
- – les cardiopathies cyanogènes s'accompagnent d'altérations maculaires et de la tête du nerf optique visibles en OCT [7] qu'il faut impérativement évaluer, car elles traduisent l'hypoxie chronique et ses retentissements anatomiques;
- – dans les cas d'hypertension artérielle (HTA) mal équilibrée, le bilan oculaire est indispensable à la recherche de lésions vasculaires.
Dans toutes les maladies génétiques précédemment citées le point d'entrée peut également être ophtalmologique, l'enfant doit alors pouvoir être référé au cardiopédiatre.
Lorsqu'une intervention chirurgicale ophtalmologique est indiquée, même en dehors de tout contexte polymalformatif, le bilan anesthésique préopératoire peut mettre en évidence des anomalies à l'examen clinique cardiaque (le plus souvent perception d'un souffle) amenant à devoir exclure une anomalie cardiaque avant l'anesthésie. Classiquement, en cas de cataracte congénitale, on recherche une myocardiopathie associée (rare syndrome de Sengers).
Certains agents médicamenteux, soit à usage diagnostique oculaire soit à usage thérapeutique oculaire, peuvent avoir des effets secondaires cardiaques. Nous citerons pour exemple les collyres dilatateurs pupillaires (atropine qui peut augmenter la fréquence cardiaque et phényléphrine qui peut augmenter la tension artérielle), les collyres β-bloquants (pouvant ralentir la fréquence cardiaque) dans le traitement du glaucome, les β-bloquants par voie systémique dans le traitement des angiomes orbitaires, etc. Ces agents médicamenteux doivent être prescrits après avis éventuel du pédiatre ou du cardiopédiatre, en particulier en cas de traitement au long cours, et a fortiori lorsque l'enfant présente une cardiopathie (congénitale, acquise) ou une arythmie.
[1] Steindl K Marfan syndrome and related connective tissue disorders Praxis (Bem 1994): ( 2013 ) : 102: 1483-1488
[2] McMain K, Robitaille J, Smith I Ocular features of CHARGE syndrome J AAPOS: ( 2008 ) : 12: 460-465
[3] Saleh M, Kamath BM, Chitayat D Alagille syndrome : clinical perspectives Appl Clin Genet: ( 2016 ) : 9: 75-82
[4] Gokturk B, Topcu-Yilmaz P, Bozkurt B Ocular findings in children with 22q11.2 deletion syndrome J Pediatr OPhthalmol Strabismus: ( 2016 ) : 53: 218-222
[5] Joyce CA, Zorich B, Pike SJ Williams-Beuren syndrome : phenotypic variability and deletions of chromosomes 7, 11, and 22 in a series of 52 patients J Med Genet: ( 1996 ) : 33: 986-992
[6] Brennan D, Giles S Ocular involvment in fetal alcohol spectrum disorder : a review Curr Pharm Des: ( 2014 ) : 20: 5377-5387
[7] de Aguiar Remigio MC, Brandt CT, Santos CCL Macular and peripapillary retinal nerve fiber layer thickness in patients with cyanotic congenital heart disease Eye: ( 2015 ) : 29: 465-468
A. Carsin, M. Callet
La relation entre le pneumopédiatre et l'ophtalmologiste s'articule essentiellement autour de deux pathologies : la sarcoïdose et l'allergie.
La sarcoïdose est une maladie granulomateuse d'origine indéterminée dont les formes pédiatriques sont rares (3 % des cas). L'incidence en pédiatrie est estimée à 0,29/100 000 enfants principalement chez les enfants de plus de 10 ans [1]. La sarcoïdose peut atteindre plusieurs organes, principalement les poumons [2].
L'atteinte respiratoire peut être asymptomatique et les signes cliniques éventuels sont : toux sèche persistante, une dyspnée et/ou douleur thoracique. Dans des cas extrêmes, la sarcoïdose évolue vers la fibrose pulmonaire. L'analyse du liquide broncho-alvéolaire montre une proportion de lymphocytes T nettement augmentée avec un ratio CD4/CD8 augmenté. L'alvéolite lymphocytaire a une spécificité diagnostique de 93 % , signe précoce qui précède la formation ganglionnaire. Elle n'a cependant pas, chez l'enfant, de corrélation avec l'activité de la maladie, ni avec son évolution ou l'efficacité des traitements.
L'atteinte ophtalmologique est présente dans près de 40 % des cas d'enfants atteints de sarcoïdose pulmonaire [3]. L'atteinte ophtalmologique est le plus souvent insidieuse et se présente essentiellement sous la forme d'une iridocyclite granulomateuse chronique, mais peut parfois atteindre le segment postérieur [4]. Les lésions observées peuvent toucher :
- – le segment antérieur : conjonctivite avec dépôts conjonctivaux chroniques pouvant être le premier signe de la maladie [5], infiltrats cornéens bilatéraux [6], uvéite antérieure (typiquement aiguë bilatérale granulomateuse synéchiante);
- – ainsi que le segment postérieur : uvéite postérieure et intermédiaire, hyalite avec atteinte du vitré antérieur et intermédiaire, atteinte choriorétinienne pouvant mimer une choriorétinopathie de Birdshot [7].
Un cas d'occlusion de branche veineuse rétinienne a été rapporté, ceci en l'absence de signes d'iridocyclite et sensible à un traitement par corticothérapie systémique [8]. L'implant intravitréen de dexaméthasone, adjoint à un traitement systémique immunomodulateur, serait associé à une amélioration de l'acuité visuelle ainsi qu'à un contrôle de l'inflammation intra-oculaire chez ces enfants atteints d'uvéite sarcoïdosique [9].
En cas de découverte d'une sarcoïdose, le pédiatre confiera donc systématiquement l'enfant à l'ophtalmologiste à la recherche d'une atteinte ophtalmologique de la sarcoïdose qui pourra s'intégrer dans une atteinte extrapulmonaire faisant partie des formes sévères de sarcoïdose. Le traitement sera alors discuté conjointement.
De la même façon, en présence de signes ophtalmologiques évoquant une sarcoïdose, l'ophtalmologiste confiera l'enfant au pédiatre qui réalisera les examens complémentaires orientés par la clinique.
L'ophtalmologiste et le pneumopédiatre sont amenés à traiter des enfants atteints de maladies à composante allergique comme l'asthme. La maladie allergique peut entraîner un asthme, une rhinite allergique. L'association à une conjonctivite allergique est fréquente.
Le pneumopédiatre réalisera des tests à la recherche de sensibilisations allergiques qui pourront orienter vers l'origine allergique des symptômes. En cas de non-réponse au traitement symptomatique (lavages oculaires au sérum frais, en association avec un traitement anti-histaminique ou antidégranulant mastocytaire en collyre ou par voie générale) ou de symptômes sévères, il est nécessaire de rechercher des diagnostics différentiels de la conjonctivite allergique : conjonctivite aiguë infectieuse virale ou bactérienne ou des formes associées de cette dernière (kératoconjonctivite vernale, instabilité lacrymale quantitative ou qualitative, kératoconjonctivite atopique, blépharoconjonctivite de contact), imposant un traitement spécifique pouvant aller jusqu'au recours aux immunosuppresseurs locaux (ciclosporine topique). L'atteinte ophtalmologique est étroitement reliée au terrain atopique de ces enfants et ces pathologies partagent des facteurs favorisants communs notamment la pollution aérienne, facteur de risque d'asthme établi et récemment incriminé dans les conjonctivites de l'enfant [10].
Aussi, en cas de conjonctivite allergique, l'ophtalmologiste recherche d'autres signes d'allergie comme une rhinite allergique (prurit nasal, anosmie, rhinorrhée, éternuement, obstruction nasale) et un asthme (signes respiratoires à l'effort ou au repos à type de toux, sifflement). En cas de signes respiratoires associés à une conjonctivite allergique (10 % des adolescents présentant une rhinoconjonctivite ont un asthme associé [11]), un bilan pneumologique est nécessaire à la recherche d'un asthme. La plupart du temps, le pneumopédiatre pourra conduire les tests allergologiques et éventuellement introduire une immunothérapie spécifique pouvant être bénéfique aux enfants sur les signes de conjonctivite allergique.
Le recours aux corticostéroïdes en nébulisation chez l'enfant asthmatique impose une rigueur spécifique à son âge et à sa morphologie faciale. L'utilisation de masques faciaux non adaptés ou mal positionnés au visage peut entraîner un dépôt de substance stéroïde au niveau du visage et des yeux [12], avec les effets secondaires que peut engendrer une exposition à la corticothérapie prolongée : particulièrement sur la cornée, le cristallin et sur la révélation d'un glaucome notamment en cas d'antécédents familiaux de glaucome.
L'association entre inhalation de corticoïdes au long cours chez les enfants asthmatiques et apparition d'une hypertonie, d'un glaucome ou d'une cataracte sous-capsulaire n'est cependant pas prouvée [13-15].
Sarcoïdose : forme pédiatrique rare, surtout responsable d’uvéites.
Allergie oculaire :
-
souvent associée à l’asthme (kératoconjonctivite) ;
iatrogénie : prudence avec les corticostéroïdes inhalés.
[1] Hoffmann AL, Milman N, Byg KE Childhood sarcoidosis in Denmark 1979-1994 : incidence, clinical features and laboratory results at presentation in 48 children Acta Paediatr: ( 2004 ) : 93: 30-36
[2] Baughman RP, Teirstein AS, Judson MA Clinical characteristics of patients in a case control study of sarcoidosis Am J Respir Crit Care Med: ( 2001 ) : 164: 1885-1889
[3] Nathan N, Marcelo P, Houdouin V Lung sarcoidosis in children : update on disease expression and management Thorax: ( 2015 ) : 70: 537-542
[4] Laghmari M, Karim A, Guedira K Uveitis in children : about 20 cases J Fr Ophtalmol: ( 2003 ) : 26: 609-613
[5] Caça I, Unlü K, Büyükbayram H, Ari S Conjunctival deposits as the first sign of systemic sarcoidosis in a pediatric patient Eur J Ophthalmol: ( 2006 ) : 16: 168-170
[6] Güngor SG, Akova YA, Küçükodük A, Baskin E Bilateral corneal infiltrates and uveitis in a pediatric patient with presumed ocular sarcoidosis Ocul Immunol Inflamm: ( 2013 ) : 21: 8-10
[7] Sullu Y, Yildiran A, Sullu Y Sarcoid uveitis simulating birdshot chorioretinopathy in a child Retin Cases Brief Rep: ( 2012 ) : 6: 7-10
[8] Momtchilova M, Pelosse B, Ngoma E, Laroche L Occlusion de branche veineuse de la rétine associée à une sarcoïdose chez l'enfant : à propos d'un cas Journal Français d'Ophtalmologie: ( 2011 ) : 34: 243-247
[9] Bratton ML, He YG, Weakley DR Dexamethasone intravitreal implant (Ozurdex) for the treatment of pediatric uveitis J AAPOS: ( 2014 ) : 18: 110-113
[10] Nucci P, Sacchi M, Pichi F Pediatric conjunctivitis and air pollution exposure : a prospective observational study Semin Ophthalmol: ( 2016 ) : 1-5: -
[11] Cibella F, Ferrante G, Cuttitta G The burden of rhinitis and rhinoconjunctivitis in adolescents Allergy Asthma Immunol Res: ( 2015 ) : 7: 44-50
[12] Harris KW, Smaldone GC Facial and ocular deposition of nebulized budesonide : effects of face mask design Chest: ( 2008 ) : 133: 482-488
[13] Alsaadi MM, Osuagwu UL, Almubrad TM Effects of inhaled fluticasone on intraocular pressure and central corneal thickness in asthmatic children without a family history of glaucoma Middle East Afr J Ophthalmol: ( 2012 ) : 19: 314-319
[14] Emin O, Fatih M, Mustafa O Evaluation impact of long-term usage of inhaled fluticasone propionate on ocular functions in children with asthma Steroids: ( 2011 ) : 76: 548-552
[15] Pelkonen A, Kari O, Selroos O Ophthalmologic findings in children with asthma receiving inhaled budesonide J Allergy Clin Immunol: ( 2008 ) : 122: 832-834
C. Rousset-Rouviére, M. Callet, V. Bautrant
L'atteinte ophtalmologique au cours des pathologies rénales est fréquente. Il existe plusieurs cadres nosologiques pour lesquels une coopération interdisciplinaire entre néphrologues pédiatres et ophtalmologistes est nécessaire.
La situation la plus fréquente est l'existence d'une maladie systémique, congénitale ou acquise, pouvant présenter une atteinte des deux organes. Sur le plan embryologique, certaines structures rénales et oculaires ont la même origine; il existe des similitudes dans les voies de développement et les structures moléculaires de ces deux organes expliquant leur atteinte simultanée dans certaines pathologies.
Le pédiatre doit connaître les manifestations oculaires des maladies rénales pour orienter le diagnostic étiologique au moment de la découverte de la pathologie rénale et assurer le dépistage de la survenue d'une atteinte oculaire spécifique au cours du suivi.
Certaines situations méritent un examen ophtalmologique en urgence afin de débuter une prise en charge spécifique. Cet examen recherche par exemple une uvéite dans les maladies inflammatoires, avant de débuter un traitement par corticoïdes.
Inversement, les ophtalmologistes doivent connaître les pathologies rénales qui peuvent se manifester par une atteinte oculaire initiale afin d'orienter les patients vers le pédiatre pour une prise en charge néphrologique.
Les autres situations nécessitant un dialogue entre néphrologues pédiatres et ophtalmologistes sont la recherche de complications oculaires de l'insuffisance rénale chronique, de l'hypertension artérielle ou du diabète, ainsi que les complications oculaires secondaires à certains traitements (par exemple les corticoïdes au long cours ou l'hydroxychloroquine : Plaquenil). Ces situations ne seront pas abordées dans ce sous-chapitre.
Certaines cellules épithéliales du rein et de l'œil sont des cellules ciliées. L'anomalie d'une des protéines impliquées dans le fonctionnement ciliaire peut être responsable d'une atteinte rénale et oculaire. L'atteinte rénale est caractérisée par une néphropathie tubulo-interstitielle chronique évoluant vers l'insuffisance rénale terminale à plus ou moins long terme. L'atteinte ophtalmologique est caractérisée par une dégénérescence tapétorétinienne ou rétinite pigmentaire. Les formes les plus sévères sont responsables d'une cécité dès les premières années de la vie, les formes les moins graves n'ont aucune traduction clinique et ne se manifestent qu'à l'électrorétinogramme. Ces pathologies sont le plus souvent de transmission autosomique récessive.
L'atteinte isolée de l'œil et du rein du fait de l'atteinte des cellules ciliées se nomme le syndrome de Senior-Loken.
L'atteinte d'autres organes présentant des cellules ciliées est possible notamment une atteinte neurologique, osseuse ou hépatique comme :
- – les néphronophtises et maladies apparentées : elles sont caractérisées par une néphropathie tubulo-intersitielle isolée (Senior-Loken) ou associée à des atteintes extrarénales multiples. À cette hétérogénéité clinique s'ajoute une hétérogénéité génétique avec près de 15 gènes impliqués : la mutation du gène NPHP1 responsable de la néphronophtise juvénile est la plus fréquente; les mutations des gènes NPHP5 et NPHP6 sont associées à une atteinte oculaire sévère notamment une dystrophie rétinienne. La recherche des atteintes extrarénales notamment oculaire permet de guider la recherche génétique;
- – le syndrome de Bardet-Biedl : il réunit une atteinte rénale tubulo-interstitielle, une rétinite pigmentaire sévère pouvant s'associer à une maculopathie en œil de bœuf, une obésité, une polydactylie post-axiale, un hypogonadisme et un retard mental; l'expression clinique est très variable et à ce jour 12 gènes ont été impliqués dans ce syndrome (BBS1 à BBS12);
- – le syndrome de Sensenbrenner ou dysplasie cranio-ectodermique : il est caractérisé par des anomalies squelettiques (thorax étroit, dolichocéphalie, craniosténose sagittale), des anomalies ectodermiques (anomalies dentaires, hypoplasie des ongles), une néphropathie tubulo-interstitielle, une fibrose hépatique et une rétinite pigmentaire; plusieurs gènes impliqués dans le transport ciliaire ont été identifiés dans cette maladie.
De nombreuses pathologies existent :
- – le syndrome rein-colobome : il est caractérisé par un colobome du nerf optique parfois associé à une microphtalmie et une hypoplasie rénale pouvant conduire à une insuffisance rénale terminale; une surdité peut être présente; des mutations dans le gène codant pour le facteur de transcription PAX2 peuvent être à l'origine de ces malformations;
- – le syndrome de Fraser : c'est une entité clinique rare qui associe une cryptophtalmie (malformation palpébrale où les paupières sont remplacées par une couche de peau fusionnant avec un œil microphtalme) et une syndactylie; les malformations rénales sont fréquemment associées; dans les formes les plus sévères, il existe une agénésie rénale bilatérale responsable d'une insuffisance rénale; on retrouve de plus une dysmorphie faciale et d'autres malformations viscérales; le gène responsable est FRAS1;
- – le syndrome branchio-oto-rénal (BOR) qui associe des anomalies des arcs branchiaux (fentes, fistules ou kystes branchiaux), des anomalies auditives (malformation du pavillon de l'oreille, orifices pré-auriculaires, surdité) et des anomalies rénales (hypodysplasie, agénésie rénale, rein polykystique); on peut retrouver une atteinte oculaire avec cataracte congénitale, microphtalmie ou malformations du segment antérieur de l'œil; les gènes BOR, EYA1, SIX1 et SIX5 sont impliqués dans cette pathologie de transmission autosomique dominante;
- – le syndrome CHARGE : il est défini par une association malformative complexe qui regroupe principalement colobome, malformations cardiaques (heart), atrésie des choanes, retard de croissance et/ou retard mental, hypoplasie génitale, anomalies des oreilles (ear) avec possible surdité; on peut retrouver l'association d'une microphtalmie, d'une atteinte neurologique et des hypodysplasies rénales; le syndrome CHARGE est le plus souvent sporadique ou se transmet selon un mode autosomique dominant;
- – les anomalies du gène SOX2 : les mutations de ce gène sont responsables d'une atteinte oculaire sévère de type microphtalmie ou anophtalmie associées à des malformations rénales, un micropénis et une atteinte neurologique; c'est une pathologie très rare avec une dizaine de cas rapportés;
- – le syndrome WAGR : il associe une tumeur de Wilms (néphroblastome), une aniridie totale ou partielle, des anomalies génito-urinaires et un déficit (retard) intellectuel; la découverte d'une aniridie chez un enfant doit attirer l'attention de l'ophtalmologiste et faire rechercher un néphroblastome si le caryotype montre une délétion 11p13 avec par conséquent « syndrome de gènes contigus » et délétion à la fois de PAX6 pour l'œil et WT1 pour le rein; le glaucome et la cataracte sont des manifestations oculaires pouvant être associées. Il s'agit d'une délétion de gènes contigus sur le chromosome 11;
- – le syndrome acro-réno-oculaire de transmission autosomique dominante : il associe des malformations oculaires (colobome du nerf optique, syndrome de Duane), rénales (malrotation, ectopie) et des extrémités (anomalie du rayon radial);
- – le syndrome de Williams-Beuren : il est causé par une microdélétion du gène de l'élastine en 7q 11 et caractérisé par une atteinte cardiaque (sténose aortique supravalvulaire le plus souvent), un retard psychomoteur, une dysmorphie faciale typique; des anomalies oculaires peuvent être retrouvées comme un strabisme ou des troubles de la réfraction, ainsi que des anomalies vasculaires comme des sténoses des artères rénales responsables d'une hypertension artérielle; une hypercalcémie néonatale peut évoluer vers une néphrocalcinose;
- – le syndrome d'Alagille : il est caractérisé par une cholestase chronique liée à une paucité des voies biliaires intra-hépatiques, une sténose des artères pulmonaires, des anomalies vertébrales et une dysplasie rénale; sur le plan ophtalmologique, on peut retrouver un embryotoxon postérieur ou une anomalie d'Axenfeld, des anomalies du nerf optique comme des drusen et une rétinopathie pigmentaire; les gènes JAG1 et NOTCH2 sont impliqués.
Plusieurs mitochondriopathies peuvent associer des anomalies rénales et oculaires :
- – déficit primaire en coenzyme Q10 : les mutations des gènes impliqués dans la synthèse du coenzyme Q10 sont responsables d'un déficit en coenzyme Q10, composant essentiel du transport des électrons dans la mitochondrie; on peut retrouver une atteinte rénale (le plus souvent il s'agit d'une hyalinose segmentaire et focale responsable d'un syndrome néphrotique cortico-résistant), une atteinte neurologique (encéphalomyopathie progressive), une atteinte ophtalmologique (atrophie optique) et une surdité de perception; la présentation clinique est hétérogène et dépend du gène impliqué; la recherche d'une atteinte ophtalmologique chez un patient présentant un syndrome néphrotique cortico-résistant orientant vers un déficit en coenzyme Q10 est primordial, car il s'agit de la seule mitochondriopathie accessible à un traitement substitutif pouvant modifier le pronostic de la maladie;
- – les cytopathies mitochondriales : elles associent des atteintes multiviscérales dont une atteinte rénale de type tubulaire proximale et une atteinte oculaire.
Différentes pathologies sont décrites :
- – le syndrome d'Alport : néphropathie familiale de l'adulte jeune, caractérisée par une glomérulopathie hématurique et protéinurique progressive (évoluant vers l'insuffisance rénale terminale à 20 ans en moyenne dans les formes les plus sévères, avec hypertension artérielle), une surdité neurosensorielle et une atteinte oculaire affectant le cristallin et l'épithélium pigmentaire; la manifestation oculaire caractéristique est un lenticône antérieur, parfois une cataracte partielle tardive et rarement des érosions cornéennes; mais les lésions rétiniennes pigmentaires sont plus fréquentes et se manifestent par des taches jaunâtres, ponctiformes dans la région périmaculaire et en périphérie; les anomalies oculaires peuvent être asymptomatiques sans conséquence sur la vision; le syndrome d'Alport est une maladie du collagène IV liée à des mutations de gènes codant l'une des trois chaînes alpha 3, 4 ou 5 exprimées dans la membrane basale glomérulaire; les mutations de COL4A5 sont associées au syndrome d'Alport lié à l'X (85 % des cas et plus sévère chez le garçon que chez la fille); les mutations de COL4A3 ou COL4A4 sont associées aux formes autosomiques;
- – le syndrome nail-patella : onycho-ostéo-dysplasie héréditaire associant une dysplasie des ongles (nail), des rotules (patella) hypoplasiques ou absentes et une dysplasie des coudes; une atteinte oculaire peut être associée (glaucome) de même qu'une atteinte auditive (surdité); une néphropathie glomérulaire est observée dans un tiers des cas; ce syndrome est transmis sur le mode autosomique dominant avec des mutations du gène LMX1B ;
- – le syndrome de Pierson : il associe un syndrome néphrotique congénital, une microcorie et un retard mental; il évolue souvent vers le décès; on peut retrouver des anomalies du cristallin, de la cornée et de la rétine; des mutations du gène LAMB2 ont été identifiées dans ce syndrome.
On regroupe sous ce terme les maladies entraînant une dysfonction tubulaire rénale responsable d'un trouble de la réabsorption tubulaire. Plusieurs anomalies innées du métabolisme ou certaines anomalies de transporteurs sont responsables d'une atteinte rénale tubulaire pouvant s'associer à des anomalies extrarénales. La précocité du diagnostic et donc du traitement est un facteur pronostic important.
On distingue :
- – la cystinose : elle est causée par l'accumulation intralysosomiale de cystine secondaire à une mutation du gène CTNS qui code pour le transporteur de la cystine (la cystinosine); elle est caractérisée par des dépôts de cystine dans différents organes; au niveau rénal l'atteinte tubulaire, complète et sévère, est responsable d'un syndrome de Fanconi (fuite urinaire de sodium, potassium, bicarbonates, phosphates, acide urique) entraînant une déshydratation chronique, un retard de croissance et un rachitisme; au niveau ophtalmologique, on retrouve une accumulation de cristaux de cystine dans la cornée, la conjonctive, la rétine, etc., caractéristiques de la maladie dès les premiers mois de vie. Sans traitement, la maladie évolue vers l'insuffisance rénale terminale et la cécité par atteinte cornéenne, essentiellement par diminution de la transparence, et s'accompagne des conséquences de l'accumulation de cystine dans d'autres organes comme le cœur. Le traitement spécifique par la cystéamine par voie générale et en collyre ophtalmologique permet de ralentir l'évolution de la maladie;
- – l' hyperoxalurie primitive : c'est un déficit enzymatique, caractérisé par une synthèse excessive et une excrétion augmentée de l'acide oxalique dans les urines, responsable d'une néphrocalcinose ou de lithiase rénale; sans prise en charge, elle évolue rapidement vers l'insuffisance rénale terminale; comme pour le rein, les tissus oculaires vascularisés sont surchargés de cristaux d'oxalate de calcium; une accumulation de cristaux blanc jaunâtre est visible sur la rétine interne au pôle postérieur; certaines lésions plus profondes deviennent pigmentées donnant un aspect de dégénérescence maculaire brune irrégulière; le traitement repose sur des mesures préventives visant à empêcher la précipitation d'oxalate dans les urines par une hyperhydratation et une administration de citrate; le seul traitement pouvant diminuer l'accumulation d'oxalate est la pyridoxine mais la réponse au traitement est inconstante;
- – le syndrome de Lowe ou syndrome oculo-cérébro-rénal : il associe une tubulopathie, une atteinte oculaire de type cataracte et glaucome précoces, avec un signe particulier qui est le myosis avec mauvaise dilatation pupillaire; on peut retrouver un lenticône postérieur et un trouble du développement neurologique; le gène impliqué est OCRL1 ;
- – la galactosémie : c'est une affection héréditaire de transmission autosomique récessive caractérisée par une accumulation excessive de galactose par défaut de conversion du galactose en glucose; la maladie se manifeste dès l'alimentation lactée par une atteinte digestive (vomissement, diarrhée, anorexie); on retrouve une atteinte hépatique (hépatosplénomégalie, insuffisance hépatocellulaire), des hypoglycémies, une tubulopathie, une atteinte oculaire (cataracte précoce « en gouttelettes d'huile » ) et un retard mental; le diagnostic repose sur le dosage sanguin de l'activité enzymatique de la galactose-1-phosphate uridyl transférase; il existe une amélioration des signes cliniques à la suppression du galactose alimentaire (parfois disparition de la cataracte).
Différentes anomalies du métabolisme associent atteinte du rein et de l'œil, en particulier la maladie de Fabry qui représente une erreur innée du métabolisme glycosphingoïde résultant de l'activité défectueuse de l'alpha-galactosidase A. L'atteinte rénale comporte une protéinurie apparaissant entre 20 et 40 ans et évolue vers l'insuffisance rénale terminale. Elle est suspectée devant des acroparesthésies, une hypohidrose, des douleurs abdominales non spécifiques et des angiokératomes cutanés. Les atteintes oculaires caractéristiques (cornea verticillata, cataracte, télangiectasies conjonctivales) peuvent apparaître dans l'enfance. Le diagnostic repose sur le dosage de l'alpha-galactosidase A dans les leucocytes et l'étude moléculaire. Le traitement par enzyme recombinante renforce l'intérêt d'un diagnostic précoce.
Plusieurs pathologies inflammatoires associent maladies de l'œil et du rein :
- – le TINU syndrome (TINU pour tubulo-interstitial nephritis and uveitis) : entité clinique rare d'étiologie inconnue qui associe une néphropathie tubulo-interstitielle aiguë et une uvéite antérieure bilatérale; il atteint le plus souvent les femmes jeunes; les manifestations oculorénales succèdent habituellement à une altération de l'état général et sont accompagnées d'un syndrome inflammatoire biologique; l'atteinte oculaire peut précéder, accompagner ou suivre l'atteinte rénale; l'uvéite doit être recherchée systématiquement au cours de toute néphrite interstitielle aiguë, car elle peut être asymptomatique. Réciproquement, l'exploration de la fonction rénale doit être pratiquée devant toute uvéite de l'enfant; le diagnostic reste un diagnostic d'élimination après avoir recherché les autres étiologies responsables de l'association d'une néphropathie tubulo-interstitielle et d'une uvéite, notamment les causes infectieuses et les maladies de système; le traitement repose sur la corticothérapie par voie générale ou locale; la néphropathie est de pronostic favorable, réversible dans la majorité des cas, mais l'uvéite tend le plus souvent à récidiver;
- – les vascularites à ANCA (anti-neutrophil cytoplasmic antibodie) dont il existre trois types : granulomatose de Wegener, polyangéite microscopique et syndrome de Churg et Strauss; les symptômes apparaissent principalement à l'adolescence mais des formes plus précoces sont possibles; l'atteinte rénale est une glomérulonéphrite nécrosante segmentaire et focale qui s'associe à différentes atteintes : voies respiratoires supérieures, poumons, peau, système nerveux, intestin, œil (kératite ulcérante périphérique, sclérite nécrosante, périartérite rétinienne occlusive);
- – la sarcoïdose : maladie de système de l'adulte jeune avec des manifestations thoraciques, oculaires, cutanées et rénales; l'atteinte oculaire la plus fréquente est l'uvéite antérieure bilatérale granulomateuse avec des nodules de Koeppe et de Busacca au niveau de l'iris; mais on peut retrouver aussi une uvéite postérieure avec des atteintes rétiniennes; au niveau rénal, on peut retrouver une néphropathie tubulo-interstitielle aiguë avec présence de granulome ou une atteinte glomérulaire;
- – le syndrome de Goodpasture : glomérulopathie auto-immune qui peut évoluer vers l'insuffisance rénale en absence de traitement, elle est associée à des symptômes multisystémiques notamment pulmonaires; l'atteinte oculaire peut être une sclérite nécrosante ou des atteintes rétinienne se manifestant par des hémorragies rétiniennes, des nodules cotonneux et un décollement de rétine exsudatif;
- – le lupus érythémateux disséminé : c'est une maladie de système dans laquelle le rein et l'œil peuvent être touchés; au niveau oculaire, on peut retrouver une kératite ulcérante périphérique, une vascularite rétinienne, une neuropathie optique et l'atteinte rénale est de type glomérulaire.
D'autres maladies inflammatoires, auto-immunes ou de système peuvent atteindre à la fois le rein et l'œil comme des pathologies infectieuses acquises (tuberculose, syphilis) ou encore des pathologies génétiques tumorales comme la sclérose tubéreuse de Bourneville ou la maladie de von Hippel-Lindau.
À travers cette liste de pathologies non exhaustive, l'importance d'une coopération entre pédiatres néphrologues et ophtalmologistes apparaît essentielle. Le diagnostic précoce d'une atteinte oculaire au cours de certaines maladies rénales peut permettre de mettre en route un traitement spécifique pouvant modifier l'évolution de la maladie. Dans d'autres cas, la recherche d'une atteinte oculaire au cours des pathologies rénales permet de poser un diagnostic étiologique.
Ciliopathies : néphronophtises, syndrome de Bardet- Biedl, syndrome de Sensenbrenner.
Malformations rénales : syndrome rein-colobome, syndrome de Fraser, syndrome branchio-oto-rénal (BOR), syndrome CHARGE, anomalies du gène SOX2, syndrome WAGR, syndrome acro-réno-oculaire, syndrome d’Alagille.
Mitochondriopathies : déficit primaire en coenzyme Q10, cytopathies mitochondriales.
Glomérulopathies congénitales : syndrome d’Alport, syndrome nail-patella, syndrome de Pierson.
Tubulopathies congénitales : cystinose, hyperoxalurie primitive, syndrome de Lowe, galactosémie.
Autres erreurs innées du métabolisme : maladie de Fabry.
Pathologies acquises inflammatoires, de système et auto-immunes : TINU syndrome, vascularites à ANCA, sarcoïdose, syndrome de Goodpasture, lupus érythémateux disséminé, infections, phacomatoses.
J. -M. Triglia, M. Callet
L'ORL et l'ophtalmologiste ont un champ médical transversal souvent mal connu : d'une part, dans les pathologies en rapport avec la surdité; d'autre part, dans les pathologies de voisinage.
Chez les enfants sourds, la place de la vision est fondamentale, du fait de la très grande dépendance visuelle de ces enfants pour communiquer. Toutes les anomalies ophtalmologiques doivent être prises en charge aussi rapidement que possible pour permettre à l'enfant de s'appuyer sur les indices visuels de la communication. Par ailleurs, il est également rapporté qu'en comparaison avec des enfants non déficients auditifs, les enfants sourds présentent davantage d'anomalies oculaires, ce qui souligne l'importance de l'examen visuel [1].
Parallèlement, l'examen ophtalmologique permet d'orienter l'enquête étiologique d'une surdité. Dans cette enquête, l'examen ophtalmologique peut retrouver des signes évocateurs de certaines causes extrinsèques (toxoplasmose, rubéole, CMV, etc.) et de certains syndromes associant surdité et troubles visuels, dont plus de cinquante sont décrits. Parmi ceux-ci, nous citerons ici les plus fréquents.
Mais de même que l'ORL demande un bilan visuel, l'ophtalmologiste doit savoir demander un bilan auditif devant des anomalies visuelles qui peuvent précéder l'apparition d'une surdité.
Le syndrome CHARGE (Coloboma, Heart defect, Atresia choanae, Retarded growth, Genital anomalies, Ear anomalies) est responsable de 16 % des causes infantiles de surdi-cécité. Il inclut colobome oculaire, anomalies cardiaques, atrésie des choanes, retard de croissance et du développement, anomalies génitales et des oreilles avec surdité. Il a été reporté sur le plan ophtalmologique l'association colobome oculaire-paralysie oculomotrice [2].
Le tableau de la trisomie 21 est bien connu, il associe : rétinopathie des prématurés [3], erreurs réfractives, nystagmus, strabisme, épiphora, atteintes de tous les segments oculaires (anomalies palpébrales, kératocône, tâches iriennes de Brushfield, cataractes, glaucome congénital, anomalies des vaisseaux rétiniens, hypoplasie fovéolaire, pâleur papillaire, anomalies pigmentaires focalisées, colobome, etc.) [4]. Les études montrent que 80 % des enfants atteints de trisomie 21 ont des capacités d'accommodation réduites [5]. Il est à noter que ces enfants présentent également dans 30 à 80 % des cas un syndrome d'apnées obstructives du sommeil pouvant être à l'origine de pathologies ophtalmologiques acquises telles que glaucome, occlusions vasculaires rétiniennes, aggravation d'une rétinopathie diabétique ultérieure [6, 7].
Ce syndrome est la cause la plus fréquente de surdité liée à une cécité. Il existe trois type de syndrome d'Usher :
- – le type 1 associe une surdité profonde bilatérale à une aréflexie vestibulaire, responsable des retards d'acquisition (retards à la tenue de la tête, à la station assise et à la marche). La rétinite pigmentaire apparaît de façon précoce en période prépubertaire (10 ans), expliquant qu'un fond d'œil normal chez le tout jeune enfant n'exclut pas une rétinopathie pigmentaire d'où le recours nécessaire à l'électrorétinogramme (ERG). Cependant, un ERG normal avant l'âge de 3 ans ne peut pas éliminer formellement le diagnostic d'Usher;
- – le type 2 se caractérise par une surdité moyenne à sévère stable et d'apparition tardive. La rétinite pigmentaire apparaît aussi tardivement, vers 20 à 30 ans avec une évolution moins sévère que dans le type 1;
- – dans le type 3, la surdité débute à la puberté; elle est évolutive et quelquefois associée à une atteinte vestibulaire. La rétinopathie pigmentaire apparaît plus tardivement et est moins sévère que dans le type 1.
Les syndromes d'Usher peuvent entraîner également d'autres anomalies ophtalmologiques telles qu'un glaucome pigmentaire [7] et/ou un colobome maculaire [8].
Sur le plan génétique, plusieurs gènes sont impliqués non seulement pour chacun des trois types, mais aussi pour un même type de syndrome d'Usher.
Le syndrome de Cockayne (retard mental, nanisme, surdité de perception qui débute dans la deuxième année de vie) peut être associé à une énophtalmie, une rétinopathie pigmentaire précoce et d'évolution rapide, une cataracte, une dégénérescence cornéenne précoce [9], ainsi qu'à une photosensibilité cutanée et conjonctivale devant faire rechercher des tumeurs précoces. L'évolution est caractérisée par l'apparition de signes de sénilité précoce.
Ce syndrome se caractérise par une obésité et un diabète sucré, une surdité neurosensorielle (légère ou modérée) lentement progressive, associée à une rétinopathie pigmentaire et à une dystrophie des cônes et des bâtonnets [10]. La cécité survient en deuxième décennie de vie avec nystagmus.
C'est une affection héréditaire dominante liée à l'X, caractérisée par l'existence d'une néphropathie hématurique progressive qui précède classiquement la surdité et l'atteinte oculaire. La surdité est précoce, prédominant sur les fréquences aiguës dans la première décennie. Progressivement, elle devient bilatérale [11]. L'atteinte oculaire est rare chez les enfants et les jeunes patients et augmente en fréquence et en sévérité avec l'âge. Les lésions caractéristiques sont le lenticône antérieur (qui est parfois une aide au diagnostic, marqueur de gravité du syndrome et souvent bilatéral), l'érosion cornéenne récidivante, l'atteinte maculaire à type de ponctuations mais aussi de trous maculaires géants [12], de rétinopathie mouchetée et de dystrophie cornéenne postérieure polymorphe [13, 14]. Il faut savoir rechercher une protéinurie microscopique en cas de décollement de la rétine chez un patient jeune au profil évocateur.
Ce syndrome associe dans 80 % des cas une surdité neurosensorielle sévère ou profonde et évolutive apparaissant tôt dans l'enfance. Cette surdité peut être également mixte du fait d'otites séromuqueuses favorisées par une possible fente palatine [11].
À côté des anomalies cartilagineuses et ligamentaires (hyperlaxité), l'atteinte ophtalmologique se manifeste par une forte myopie, des anomalies du vitré et un risque élevé de décollement de rétine (60 % des patients) conduisant à une cécité chez 4 % des patients [15].
C'est une pathologie génétique grave associant un diabète [16], une atrophie optique primitive bilatérale, une surdité neurosensorielle bilatérale et symétrique évolutive affectant au début les fréquences aiguës. Elle débute entre la première et la deuxième décennie. Elle implique en pratique la réalisation d'un bilan glycémique en cas d'atrophie optique.
La collaboration entre ophtalmologiste et ORL se justifie également en présence de symptômes tels qu'une instabilité et des vertiges. En l'absence d'anomalies vestibulaires et neurologiques caractérisées, une pathologie oculaire doit être recherchée, les principales anomalies touchent la réfraction et les vergences oculaires. La prise en charge de ces anomalies corrige les symptômes. Ainsi, le bilan ophtalmologique devra comprendre pour ces enfants l'étude de l'acuité visuelle, la réfraction sous cycloplégique, l'étude de la motricité oculaire intrinsèque et extrinsèque, en y associant un fond d'œil systématique.
Le voisinage anatomique entre les régions relevant de l'ophtalmologie et celles relevant de l'ORL expliquent aisément l'intrication et le nombre important de symptômes et d'affections communs aux deux spécialités.
De diagnostic aisé lorsqu'il est effectué au cours de la période anténatale, le kyste lacrymonasal est plus difficile à reconnaître après la naissance. Il peut être à l'origine d'une détresse respiratoire néonatale en cas de forme bilatérale ou quasiment asymptomatique dans les formes unilatérales. Une dacryocystite en période néonatale en est un symptôme clé. Le kyste est vu en endoscopie nasale. Le traitement curatif repose sur une marsupialisation du kyste, des collyres antibiotiques et un massage régulier du sac lacrymal (encadré 27-2).
Prise en charge ophtalmologique et ORL d’une distension néonatale du sac lacrymal
CLINIQUE
Forme simple : dacryocystocèle
Distension médio-canthale constatée dès la naissance, située sous le tendon canthal médial de coloration bleutée pseudo-angiomateuse.
Palpation digitale : caractère liquidien rénitent sans aucun reflux de mucus par les canalicules.
Examen rhinoscopique antérieur (s’il est réalisé) : protrusion qui comble le méat nasal inférieur sous le cornet inférieur, plus ou moins médialisé.
Attention aux rares formes bilatérales et extensives pouvant entraîner une détresse respiratoire néonatale ; elles peuvent être dépistées par l’échographie in utero afin d’établir préventivement un plan de soins adapté.
Forme compliquée de dacryocystite (fig. 27-2a et b)
Surinfection (20 % des cas) du contenu lacrymal avec remplacement de la coloration bleutée par une couleur rouge.
Sécrétions purulentes conjonctivales.
Absence de fièvre.
Évolution : favorable (guérison spontanée dans 80 % des cas et au cours du premier mois), pronostic esthétique excellent, fistulisation sacculo-cutanée exceptionnelle.
Ce que l’ORL doit rechercher dans les deux formes
Masse intranasale visible sous le cornet inférieur qu’elle soulève vers le haut.
Consistance molle, d’allure kystique, translucide ou de couleur jaunâtre, blanc nacré ou bleutée.
TRAITEMENT
Traitement local (dacryocystocèle + dacryocystite)
Massage du sac lacrymal matin et soir.
Lavage oculaire et des fosses nasales au sérum physiologique 2 fois/jour.
Collyre local : Azyter® 2 fois/jour pendant 3 jours (prévention de la surinfection en cas de dacryocystocèle).
Traitement général (dacryocystite)
Antibiothérapie à large spectre (Haemophilus et streptocoque) : amoxicilline-acide clavulanique (Augmentin®), 1 dose/poids 3 fois/ jour pendant 5 jours.
Traitement chirurgical
Traitement du kyste : exérèse-marsupialisation du kyste au laser ou aux ciseaux sous anesthésie générale (fig. 27-2c et d).
Sondage des voies lacrymales à distance de l’épisode infectieux.
Traitement à réaliser
En semi-urgence pour les formes unilatérales.
En urgence pour les formes bilatérales (32 % des cas) responsables de détresse respiratoire.
ÉVOLUTION
Guérison postopératoire (fig. 27-2e et f).
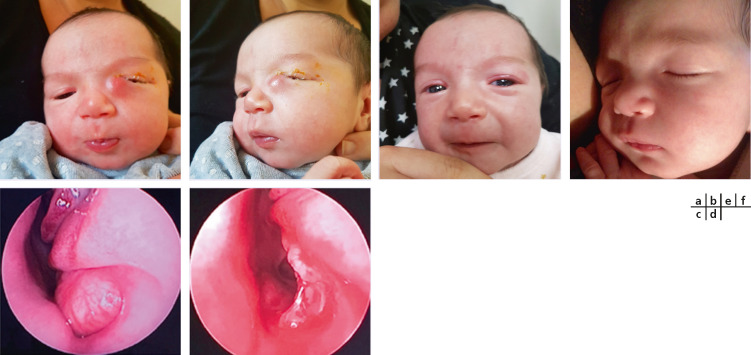
Fig. 27-2 Distension néonatale du sac lacrymal.
a, b. Nouveau-né de 9 jours présentant une dacryocystite aiguë sur dacryocystocèle gauche : face (a), profi l (b). c, d. Visualisation endoscopique d’un kyste lacrymonasal de la fosse nasale gauche (c) et traitement chirurgical après marsupialisation (d). e, f. Nouveau-né en postopératoire à 18 jours : disparition de la dacryocystocèle gauche compliquée de dacryocystite après marsupialisation : face (e), profi l (f).
Les tumeurs de la fosse nasale ou des sinus, qu'elles soient bénignes ou malignes, peuvent comprimer ou envahir les voies lacrymales et être responsables d'un larmoiement ou de dacryocystites.
Les dacryocystites constituent un diagnostic différentiel habituel des ethmoïdites aiguës et parfois, seule la tomodensitométrie peut les différencier. Les sinusites aiguës peuvent être à l'origine de complications orbitaires par diffusion du processus infectieux, essentiellement quand il s'agit des ethmoïdites aiguës; elles peuvent être à l'origine d'abcès sous-périostés, de cellulites intra-orbitaires, d'abcès intra-orbitaires, voire de thrombose du sinus caverneux ou de cécité. Un bilan de l'oculomotricité et une évaluation de l'acuité visuelle conditionnent la réalisation d'une imagerie et d'un éventuel drainage chirurgical en urgence. Les sphénoïdites peuvent s'accompagner d'une baisse de l'acuité visuelle ou de paralysies oculomotrices.
Au décours d'une chirurgie sinusienne, toute modification clinique ophtalmologique doit motiver une consultation ophtalmologique urgente à la recherche d'une lésion iatrogène.
Les pulvérisations nasales de corticoïdes, dans le cadre d'une rhinosinusite chronique ou d'une polypose nasale ou en postopératoire de chirurgie ORL, ne semblent pas avoir d'effet délétère sur les segments antérieur et postérieur. Elles ne sont pas responsables d'une augmentation de la pression intra-oculaire chez les enfants [17, 18].
La collaboration entre l’ophtalmologiste et l’ORL pédiatre concerne surtout :
la surdité :
-
entrant dans le cadre de nombreux syndromes, la surdité peut s’associer à une atteinte ophtalmologique pouvant intéresser toutes les structures de l’oeil,
le syndrome d’Usher associe surdité et rétinopathie pigmentaire ;
les pathologies de voisinage, notamment sinusiennes pouvant avoir un effet direct sur l’oeil via la proximité anatomique par un mécanisme d’envahissement ou de diffusion.
[1] Ostadimoghaddam H, Mirhajian H, Yekta AY Eye problems in children with hearing impairment J Curr Ophthalmol: ( 2015 ) : 27: 56-59
[2] Yang HK, Choi BY, Kim JH CHARGE syndrome with oculomotor nerve palsy J AAPOS: ( 2015 ) : 19: 555-557
[3] Movsas TZ, Spitzer AR, Gewolb IH Trisomy 21 and risk of retinopathy of prematurity Pediatrics: ( 2015 ) : 136: e441-e447
[4] Copin H, Brémont-Gignac D Manifestations oculaires de la trisomie 21 et aspects cytogénétiques J Fr Ophtalmol: ( 2004 ) : 27: 958-959
[5] Adyanthaya R, Isenor S, Muthusamy B Children with Down syndrome benefit from bifocals as evidenced by increased compliance with spectacle wear J AAPOS: ( 2014 ) : 18: 481-484
[6] Stores RJ, Stores G The significance of aspects of screening for obstructive sleep apnoea in children with Down syndrome J Intellect Disabil Res: ( 2014 ) : 58: 381-392
[7] Leroux les Jardins G, Glacet-Bernarda A, Lasryb S Occlusion veineuse rétinienne et syndrome d'apnée du sommeil J Fr Ophtalmol: ( 2009 ) : 32: 420-424
[8] Koucheki B, Jalali KH Pigmentary glaucoma accompanied by Usher syndrome J Glaucoma: ( 2012 ) : 21: 392-393
[9] Wu Y, Zheng Y, Yan X Ocular findings in a patient with Cockayne syndrome with two mutations in the ERCC6 gene Ophthalmic Genet: ( 2016 ) : 17: 1-3
[10] Ahmad A, D'Souza B, Yadav C Metabolic Syndrome in childhood : rare case of alstrom syndrome with blindness Indian J Clin Biochem: ( 2016 ) : 31: 480-482
[11] Triglia JM. Site Internet du service ORL Pédiatrique CHU Timone APHM. En ligne : www.orl-marseille.com
[12] Mercé E, Korobelnik JF, Delyfer MN, Rougier MB Un nouveau cas de trou maculaire géant chez un patient présentant un syndrome d'Alport J Fr Ophtalmol: ( 2012 ) : 35: 573-579
[13] Hentati D, Sellami K, Makni M Atteinte oculaire au cours du syndrome d'Alport. À propos de 32 cas J Fr Ophtalmol: ( 2008 ) : 31: 597-604
[14] Blaise P, Delanaye P, Martalo O Le lenticône antérieur : aide diagnostique au syndrome d'Alport J Fr Ophtalmol: ( 2003 ) : 26: 1075-1082
[15] Couchouron T, Masson C Le syndrome de Stickler Revue du Rhumatisme: ( 2010 ) : 77: 458-462
[16] Conart JB, Maalouf T, Jonveaux P Le syndrome de Wolfram : mise au point clinique et génétique à propos du cas de deux sœurs J Fr Ophtalmol: ( 2011 ) : 34: 543-546
[17] Seiberling KA, Chang DF, Nyirady J Effect of intranasal budesonide irrigations on intraocular pressure Int Forum Allergy Rhinol: ( 2013 ) : 3: 704-707
[18] Ozkaya E, Ozsutcu M, Mete F Lack of ocular side effects after 2 years of topical steroids for allergic rhinitis J Pediatr Ophthalmol Strabismus: ( 2011 ) : 48: 311-317
M. Callet, L. Guyot, F. Cheynet, N. Levy, D. Denis
La coopération interdisciplinaire entre l'ophtalmologiste et le chirurgien maxillofacial est particulièrement importante dans deux situations : les fractures du plancher de l'orbite et la cellulite orbitaire.
Ces deux situations nécessitent une prise en charge en urgence, seule garante d'une guérison sans séquelle ophtalmologique.
Deux fiches pratiques sont rédigées :
- – prise en charge ophtalmologique et maxillofaciale d'une fracture du plancher de l'orbite chez l'enfant (encadré 27-3) [1, 2];
- – prise en charge ophtalmologique et maxillofaciale d'une cellulite orbitaire de l'enfant (encadré 27-4) [3, 4].
Prise en charge ophtalmologique et maxillofaciale d’une fracture du plancher de l’orbite chez l’enfant
Clinique
Les fractures de l’orbite représentent 30 % des fractures du massif facial chez l’enfant, avec un pic de fréquence vers 9-10 ans et une prépondérance masculine.
À évoquer devant tout traumatisme orbitofacial.
Types de fracture
En trappe (très fréquente) :
-
sans incarcération ;
avec incarcération musculaire (droit interne, droit inférieur, petit oblique) ;
avec incarcération graisseuse.
En clapet avec effondrement du plancher (minime, modéré, majeur).
Examen ophtalmologique
Signes cliniques : baisse d’acuité visuelle, oedème palpébral, hémorragie sous-conjonctivale, plaie pénétrante du globe oculaire.
Examens : champ visuel au doigt, étude de la mobilité oculaire (primordial à la recherche de limitation douloureuse), anesthésie de la région du nerf infra-orbitaire.
Examen maxillofacial complet
Recherche fracture orbitozygomatique, fracture de la base du crâne, fracture du toit de l’orbite ou dento-alvéolaire, trouble de l’occlusion.
Examen pédiatrique général
Signes vagaux (bradycardie, malaise, pâleur) ou agitation : signe d’incarcération.
Signes neurologiques : recherche rhinorrhée, anosmie évoquant une fracture de la base du crâne avec risque méningé.
Lésions associées (polytraumatisme) : des membres, des cavités (thorax, abdomen, bassin).
Traitement


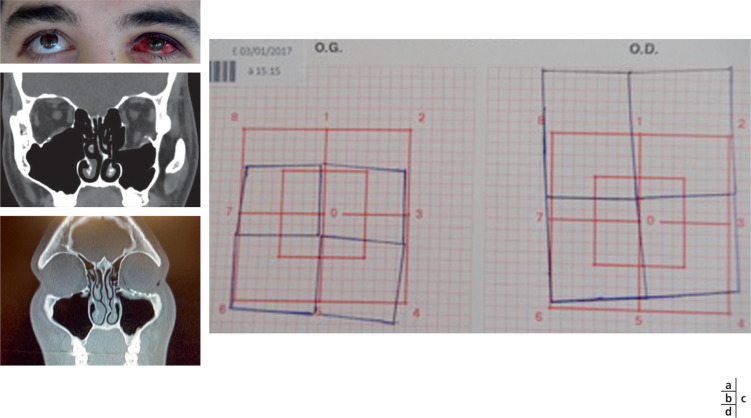
Fig. 27-3 Fracture du plancher de l’orbite gauche.
a. Patient de 18 ans consultant aux urgences pour contusion de l’oeil gauche (traumatisme par coup de poing). Pas d’antécédent, pas de traitement. Acuité visuelle de loin sans correction 10/10 et 7/10 non améliorable (ulcère cornéen central), hémorragie sous-conjonctivale diffuse, ecchymose péri-orbitaire, oedème palpébral supérieur, énophtalmie gauche sans défi cit sensitif dans le territoire du V2. Fond d’oeil de l’oeil gauche : pôle postérieur normal, contusion rétinienne à 2 heures, rétine à plat sur 360°. Oculomotricité au doigt : limitation à l’élévation de l’oeil gauche avec diplopie binoculaire. b. TDM massif facial, coupe coronale : fracture du plancher de l’orbite de l’oeil gauche avec effondrement sans incarcération musculograisseuse. c. Test de Hess-Lancaster : défi cit de l’élévation de l’oeil gauche avec hyperaction controlatérale droite. Le patient est pris en charge en urgence en chirurgie maxillofaciale ; méthylprednisolone 2 mg/kg/24 h et traitement local. d. TDM massif facial, coupe coronale : après intervention de libération et mise en place d’un implant.
Surveillance
Postopératoire : acuité visuelle, signes d’hématome compressif (exophtalmie brutale et douloureuse) =
-
soit traitement conservateur : absence de diplopie, d’incarcération pour éventuelle chirurgie ;
soit indication opératoire « à froid » : absence d’amélioration, aggravation.
Séquelles :
-
cicatricielles : énophtalmie, rétraction palpébrale, ectropion, entropion ;
oculomotrices : diplopie séquellaire (surtout si prise en charge thérapeutique > 12 h).
Prise en charge ophtalmologique et maxillofaciale d’une cellulite orbitaire de l’enfant
Clinique
Recherche de la porte d’entrée
Ophtalmologique : dacryocystite.
ORL et dentaire : ethmoïdite (dès la naissance), sinusite maxillaire (dès 3 ans), sinusite frontale (5-10 ans), sinusite sphénoïdale (10-15 ans).
Cutanée : infection locorégionale, traumatisme ± corps étranger, morsure, chirurgie.
Classification en 5 stades de Chandler
Stade I – cellulite préseptale : oedème inflammatoire palpébral.
Stades II à V avec atteinte sévère :
-
cellulite orbitaire : oedème orbitaire diffus (stade II) ;
abcès orbitaire sous-périosté (stade III) ;
abcès orbitaire (stade IV) ;
thrombose du sinus caverneux (stade V).
Signes de gravité à rechercher
Locaux : baisse d’acuité visuelle, trouble de l’oculomotricité voire ophtalmoplégie, chémosis, mydriase, anesthésie cornéenne, exophtalmie avec exposition cornéenne.
Généraux : sepsis, atteinte méningée, troubles du comportement ou troubles neurologiques, hyperleucocytose, CRP élevée.
Imagerie
Radiographies simples des sinus : non indiquée même si possibilité d’objectiver une opacité sinusienne.
TDM orbitaire sans et avec injection de produit de contraste permettant d’objectiver le stade de la pathologie (selon Chandler) et : de rechercher des signes de gravité tels qu’un abcès ; de localiser et préciser la taille d’une lésion orbitaire ; de vérifier l’état des sinus de la face ; de rechercher la présence de bulles d’air, d’une thrombose du sinus caverneux, d’abcès extraduraux, d’un empyème, d’une méningite ou d’une ostéite.
IRM orbitaire : cas complexes, complication intracrânienne ou encore dissociation entre tableau clinique évocateur et TDM normale.
Traitement

C3G : céphalosporine de 3e génération ; CRP : C-reactive proteine ; GPIP : Groupe de pathologie infectieuse pédiatrique ; SARM : Staphylococcus aureus résistant à la méticilline ; TDM : tomodensitométrie.
[1] Vazquez MP, Kadlub N, Soupre V Facial trauma and injury in children Ann Chir Plast Esthet: ( 2016 ) : 61: 543-559
[2] Guyot L, Lari N, Benso C Les fractures de l'orbite de l'enfant J Fr ophtalmol: ( 2011 ) : 34: 265-274
[3] Boivin L, Adenis JP Infections orbitaires de l'enfant : clinique, imagerie et traitement J Fr Ophtalmol: ( 2009 ) : 32: 368-373
[4] Le TD, Liu ES, Adatia FA The effect of adding orbital computed tomography findings to the Chandler criteria for classifying pediatric orbital cellulitis in predicting which patients will require surgical intervention J AAPOS: ( 2014 ) : 18: 271-277
G. Pech-Gourg, D. Denis
Parce que la tête des nourrissons est la partie de leur anatomie qu'ils exposent le plus au regard des adultes, les déformations crâniennes constituent un motif d'inquiétude fréquent pour leurs parents. Aucune bosse, dépression ou asymétrie n'est négligée sur le petit crâne chauve d'un nourrisson placé sur des genoux. S'il reste capital pour le médecin d'écarter précocement la suspicion d'une craniosténose vraie en connaissant les différents morphotypes cliniques caractéristiques (fig. 27-4 et voir fig. 6-14), il est aussi important de pouvoir confirmer le caractère positionnel d'une déformation crânienne et de savoir proposer précocement les mesures de correction.
La déformation crânienne posturale postérieure est une déformation acquise de la voÛte crânienne du nourrisson, soumise à des pressions extérieures sans anomalie des sutures. En fonction des modalités de pression, elle peut être unilatérale (le plus souvent, on parle alors de « plagiocéphalie positionnelle » ) ou bilatérale donnant un aspect d'arrière-crâne plat (on peut alors parler de « brachycéphalie positionnelle » ).
L'histoire de la déformation postérieure positionnelle est intimement liée à la lutte contre le syndrome de mort subite du nourrisson. L'incidence de ces déformations a connu une augmentation sans précédent lors de la mise en évidence de l'effet protecteur du décubitus dorsal pour lutter contre ce fléau en 1992. Dès lors, les sociétés savantes de pédiatrie ont généralisé la campagne américaine « back to sleep » .
S'il est important de rassurer les parents quant au pronostic neurologique et fonctionnel, donner l'impression que l'on néglige leur angoisse devant ces anomalies a pour conséquence de les jeter dans les bras des ostéopathes ou de confectionneurs d'orthèses crâniennes onéreuses.
Le diagnostic de déformation positionnelle est clinique et ne nécessite aucun examen radiologique. La déformation en règle générale devient évidente au 2 mois et s'aggrave progressivement pendant la première année en l'absence d'une prise en charge adaptée.
L'inspection de la tête et du cou recherche une inclinaison latérale de la tête témoignant d'un torticolis du fait d'une attitude compensatrice du chef ou d'une anomalie de la charnière craniovertébrale. Ce torticolis directement lié à la déformation doit être recherché attentivement; il reste sous-évalué. La mesure du périmètre crânien est systématique et reste dans des valeurs normales. La palpation des sutures permet de mobiliser les plaques osseuses les unes par rapport aux autres.
L'examen en vue supérieure est la clé du diagnostic permettant d'évaluer de façon globale la morphologie crânienne et d'affirmer le caractère positionnel de la déformation : le crâne est déformé en parallélogramme, l'aplatissement occipital est associé à une avancée homolatérale de l'oreille et de la bosse frontale ainsi qu'à une bosse pariétale compensatrice controlatérale (voir fig. 6-14). La distance tragus-canthus externe est diminuée du côté atteint témoignant de l'avancée du rocher homolatéral. A contrario, lors de craniosténoses vraies, la bosse frontale homolatérale est en retrait, alors que la bosse frontale controlatérale est avancée.
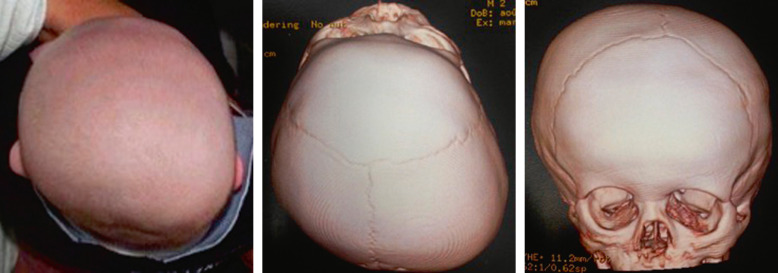
Fig. 27-4 Différents morphotypes de la déformation crânienne.
Le bilan ophtalmologique consiste en :
- – une appréciation du comportement visuel;
- – une recherche de troubles réfractifs (plus fréquents que dans la population générale);
- – un examen de l'oculomotricité notamment pour éliminer une cause oculaire lorsqu'il y a un torticolis associé à la déformation crânienne, car c'est celle-ci qui génère le torticolis lorsqu'elle est sévère (et c'est important car l'indication thérapeutique éventuelle dépend de ce torticolis « neurochirurgical » );
- – un examen du segment antérieur et un examen du fond d'œil (FO) systématique.
Gupta en 2003 [1] a étudié une population de plagiocéphalies déformationnelles (n = 111) et de plagiocéphalies synostotiques non syndromiques (n = 45), d'âge moyen 13 mois (SD = 22 mois; 0-14 ans). Il montre que les enfants atteints de plagiocéphalie déformationnelle n'ont pas plus de strabisme par rapport à la population générale, mais présentent plus souvent un astigmatisme, tandis que les enfants atteints de plagiocéphalie synostotique non syndromique présentent plus souvent un strabisme et un astigmatisme. L'astigmatisme en cas de plagiocéphalie déformationnelle est unilatéral dans 9 % des cas et bilatéral dans 15 % des cas (moyenne = 1,82 D); l'astigmatisme en cas de plagiocéphalie synostotique non syndromique est unilatéral dans 7 % des cas et bilatéral dans 21 % des cas (moyenne = 1,83 D). Sur le plan oculomoteur, le strabisme est présent dans 1 % des cas de plagiocéphalie déformationnelle et dans 7 % des cas de plagiocéphalie synostotique non syndromique. Cette étude souligne face aux déformations crâniennes la nécessité de dépister et corriger précocement les troubles réfractifs.
Par ailleurs, l'examen anatomique oculaire demeure quant à lui indispensable, du fait de la fréquence d'anomalies associées, comme des anomalies du segment antérieur : 1 cas de lenticône et 1 cas de colobome irien sont retrouvés dans la série marseillaise d'Alessi et al. sur 19 patients présentant une plagiocéphalie positionnelle [2]; à noter dans cette série 6 cas de torticolis, soit 31 % .
La prévention est sÛrement le domaine qu'il reste à développer. Tout praticien au contact d'une femme enceinte ou d'un nourrisson devrait connaître les facteurs de risque de déformation crânienne afin de sensibiliser les parents : présence d'un torticolis congénital, position préférentielle de la tête, sexe masculin, décubitus dorsal strict prolongé, première grossesse, grossesse multiple, méthode d'alimentation, hypotonie, prématurité et macrocéphalie. Il faut éduquer les parents sur la nécessité de mobiliser leur enfant. Il est également d'un intérêt majeur de réhabiliter auprès des familles le décubitus ventral lors des périodes d'éveil. La position allongée sur le ventre accélère les acquisitions psychomotrices et tonifie la musculature cervicale postérieure. Ces muscles insérés sur l'écaille occipitale vont lutter par traction contre l'aplatissement occipital.
Tout enfant en plein développement visuel doit être examiné sur le plan ophtalmologique (réfraction sous cycloplégique et FO) afin de dépister des facteurs amblyogènes. Une déformation crânienne est une situation pathologique qui favorise la survenue de ces facteurs de risque et doit donc amener le praticien à renforcer sa vigilance dans le dépistage.
Ainsi la recherche et la prise en charge de troubles réfractifs et de strabisme permettent de ne pas ajouter une déficience visuelle par amblyopie fonctionnelle dans cette population d'enfants. Cette déficience visuelle est réversible si le traitement est précoce et adapté.
L'évolution spontanée de la déformation se fait vers la normalisation de l'arrière du crâne à l'âge de 2 ans, en dehors des formes associées à un torticolis s'il n'est pas traité. En France, le traitement de référence associe des conseils de positionnement du bébé et des séances de kinésithérapie pour améliorer la mobilité de la tête et du cou. La prescription d'un casque de remodelage crânien est sujette à débat et seuls deux services de neurochirurgie sur le territoire français continuent à les prescrire. D'un point de vue scientifique, il n'y a aucune étude de qualité qui justifie son utilisation comparée à l'attitude classique de rééducation posturale.
La correction optique totale avec traitement préventif ou curatif de l'amblyopie fonctionnelle participera à la diminution du pourcentage de déficience visuelle chez les enfants.
La plagiocéphalie positionnelle n'est pas un mal nécessaire pour lutter contre la mort subite du nourrisson. Elle n'entraîne pas de trouble des apprentissages. Son traitement débute avant la naissance par l'éducation des parents sur la gestion des positions du bébé. Le dépistage et le traitement du torticolis sont essentiels.
La plagiocéphalie positionnelle peut entraîner une amblyopie qui doit être recherchée et traitée. La correction des troubles réfractifs est indispensable.
[1] Gupta PC, Foster J, Crowe S Ophthalmologic findings in patients with nonsyndromic plagiocephaly J Craniofac Surg: ( 2003 ) : 14: 522-529
[2] Alessi G, Dauletbekov D, Girard N, et al. Anomalies ophtalmologiques et radiologiques associées au plagiocéphalies postérieures positionnelles. Communication orale au 113e Congrès de la Société française d’ophtalmologie, le 06/05/2007
M. Viellard, A. Aziz-Alessi , D. Denis
Certains troubles mentaux de l'enfant s'accompagnent de troubles visuels plus fréquemment que dans la population générale. L'ophtalmologiste joue un rôle important dans le dépistage de ces troubles, dans le but d'éviter la survenue d'un surhandicap. Par ailleurs, les troubles visuels peuvent avoir d'importantes répercussions psychologiques chez l'enfant et l'adolescent : le pédopsychiatre peut être sollicité pour le dépistage et la prise en charge de troubles psychiques chez ces jeunes patients. La collaboration entre ophtalmologiste et pédopsychiatre est donc capitale dans de nombreuses situations.
Le pédopsychiatre sollicite régulièrement l'ophtalmologiste pour le dépistage de troubles de la vision chez les enfants présentant des troubles spécifiques des apprentissages, mais aussi dans le cadre d'autres troubles rencontrés classiquement en pédopsychiatrie comme l'autisme. Les troubles du spectre de l'autisme sont des troubles neurodéveloppementaux caractérisés par une altération des interactions sociales et de la communication, des centres d'intérêt et des comportements répétitifs et stéréotypés. Leur origine reste mal connue, probablement plurifactorielle, mettant en jeu des facteurs génétiques, environnementaux, épigénétiques. La Haute Autorité de santé (HAS) [1] rapporte une prévalence de la déficience visuelle chez les personnes avec autisme variant de 0 à 11,1 % selon les études. Même lorsque l'enfant avec autisme ne présente pas de déficience visuelle grave, la consultation en ophtalmologie est importante et la HAS [2] préconise depuis 2005 un examen systématique de la vision chez ces enfants. En effet, plusieurs études montrent qu'ils présentent plus fréquemment que la population générale des anomalies de la réfraction, en particulier un astigmatisme [3, 4]. Il est essentiel de dépister ces anomalies de réfraction qui peuvent entraîner un surhandicap. La mesure de l'acuité visuelle est cependant compliquée chez les enfants avec autisme. En effet, leur participation lors des examens est difficile à obtenir du fait de leurs difficultés de communication (expression et compréhension), de leurs difficultés relationnelles et de la déficience intellectuelle assez fréquemment associée à l'autisme. La situation de consultation en elle-même peut être mal vécue par ces enfants, pour qui toute situation nouvelle est source d'anxiété. La collaboration entre équipes d'ophtalmologie et de pédopsychiatrie est capitale pour préparer la consultation et faciliter la réalisation des examens : familiarisation avec la salle d'examen, délai d'attente réduit au minimum, utilisation des moyens habituels de communication de l'enfant lors des examens (pictogrammes, photos, méthode d'appariement avec préparation à la maison, etc.).
La vision joue un rôle important dans le développement dès les premiers mois de vie, et le contact œil à œil est primordial dans la construction du lien d'attachement entre l'enfant et ses parents. La déficience visuelle a donc un impact non négligeable sur le développement psychomoteur, langagier et sur l'organisation psychique du bébé. Elle perturbe l'intégration des repères du monde environnant qui constituent les bases de la pensée et de la personnalité. Elle a également un impact sur les parents, qui doivent être soutenus et guidés pour s'adapter au mieux aux besoins spécifiques de leur enfant. Certains enfants atteints de déficience visuelle présentent au cours de leur développement des comportements proches des symptômes autistiques sans être autistes pour autant. Dans d'autres cas, l'autisme est une réelle comorbidité de la déficience visuelle. La prévalence de l'autisme et de « traits autistiques » dans la population des enfants ayant une déficience visuelle varie de 0 à 53,3 % en fonction des études [1]. Ces résultats sont difficiles à interpréter du fait de l'hétérogénéité des études et de la difficulté à poser un diagnostic d'autisme chez les enfants présentant une déficience visuelle. En effet, les outils utilisés classiquement pour le diagnostic de l'autisme dépendent beaucoup de la vision et ne sont pas adaptés aux enfants ayant une déficience visuelle sévère. Les prises en charge proposées habituellement dans l'autisme sont également fondées sur des pictogrammes ou des gestes visant à pallier les difficultés langagières de ces enfants et ne sont pas adaptées en cas de déficience visuelle. L'ophtalmologiste sollicite le pédopsychiatre pour dépister les troubles autistiques chez les enfants déficients visuels. Le partenariat entre spécialistes de l'autisme et de la déficience visuelle est là encore essentiel pour élaborer des procédures d'évaluation diagnostique et de prise en charge tenant compte du handicap visuel.
L'ophtalmologiste peut être amené à adresser au pédopsychiatre ses jeunes patients dans d'autres circonstances. Les enfants et adolescents ayant une cécité congénitale semblent présenter un niveau d'anxiété supérieur à leurs pairs [5]. Lorsque la malvoyance survient plus tardivement dans l'enfance ou à l'adolescence, par exemple dans le cadre de certaines maculopathies du sujet jeune, les répercussions psychologiques sont importantes. Dans le cadre de l'onco-ophtalmologie, les enfants atteints notamment de rétinoblastome doivent être suivis sur le plan psychologique : plusieurs études ont mis en évidence les conséquences de la chimiothérapie voire des chirurgies d'énucléation chez ces jeunes enfants et leurs parents [6]. Dans un contexte de traumatisme oculaire sévère avec perte fonctionnelle voire anatomique du globe, un soutien psychiatrique de l'enfant doit absolument être réalisé du fait du fort impact sur la qualité de vie engendré par l'accident [7]. Les changements amenés par la maladie peuvent induire une rupture avec la vie antérieure de l'enfant, avec un retentissement sur la dynamique familiale [8], la vie sociale et scolaire. Il est essentiel de proposer un soutien psychologique à l'enfant et à sa famille, en particulier au moment du diagnostic.
C'est le cas du strabisme qui touche 4 % des enfants et peut avoir des répercussions importantes sur les acquisitions scolaires (la lecture en particulier), mais aussi sur les loisirs (regarder la télévision, participer à des jeux d'extérieur, etc.). Le strabisme peut également avoir un impact psychosocial négatif chez l'enfant d'âge scolaire, avec des conséquences en termes d'intégration sociale et d'estime de soi. Certaines études montrent que dès l'âge de 6 ans, les enfants ont une perception négative de leurs pairs présentant un strabisme et peuvent hésiter à les intégrer dans leurs jeux [9, 10]. En 2009, Chai [11] a mis en évidence des symptômes d'anxiété et de dépression plus importants et une moins bonne qualité de vie dans une population d'enfants de moins de 15 ans avec un strabisme que dans un groupe contrôle apparié sur l'âge et le sexe.
Ces études soulignent l'importance de dépister systématiquement et de prendre en charge les troubles émotionnels associés aux troubles de la vision rencontrés chez l'enfant et l'adolescent.
C'est également le cas des déficiences visuelles non organiques dont l'origine psychogène nécessite l'orientation vers un pédopsychiatre une fois le caractère organique écarté (voir chapitre 24.1).
L’ophtalmologiste joue un rôle important dans le dépistage de troubles visuels chez les enfants présentant divers troubles rencontrés en pédopsychiatrie, comme les troubles spécifiques des apprentissages et les troubles du spectre autistique.
Le pédopsychiatre est sollicité pour évaluer et prendre en charge les répercussions psychiques de certains troubles de la vision chez l’enfant.
Un accompagnement psychologique doit également être proposé à l’entourage de l’enfant, en particulier en cas de trouble de la vision ayant des conséquences importantes sur le fonctionnement familial.
La baisse de la vision non organique, d’origine psychogène, est un trouble somatoforme qui peut se révéler complexe, nécessitant une collaboration particulièrement étroite entre ophtalmologiste et pédopsychiatre.
[1] Haute Autorité de santé (HAS). Autisme et autres troubles envahissants du développement – état des connaissances. HAS ; 2010. En ligne : http://www.has-sante.fr/portail/jcms/c_935617/fr/autisme-et-autres-troubles-envahissants-du-developpement
[2] Haute Autorité de santé (HAS). Recommandations pour la pratique professionnelle du diag nostic de l’autisme. HAS ; 2005. En ligne : http://www.has-sante.fr/portail/jcms/c_468812/fr/recommandations-pour-la-pratique-professionnelle-du-diagnostic-de-l-autisme
[3] Denis D, Burillon C, Livet MO Ophtalmologic signs in children with autism J Fr Ophtalmol: ( 1997 ) : 20: 103-110
[4] Ezegwui IR, Lawrence L, Aghaji AE Refractive errors in children with autism in a developing country Niger J Clin Pract: ( 2014 ) : 17: 467-470
[5] Bolat N, Dogangün B, Yavuz M Depression and anxiety levels and self-concept characteristics of adolescents with congenital complete visual impairment Turk Psikiyatri Derg: ( 2011 ) : 22: 77-82
[6] Soliman SE, Dimaras H, Souka AA Socioeconomic and psychological impact of treatment for unilateral intraocular retinoblastoma J Fr Ophtalmol: ( 2015 ) : 38: 550-558
[7] Karaman S, Ozkan B, Gok M Effect of eye trauma on mental health and quality of life in children and adolescents Int Ophthalmol: ( 2016 ) - Jul 22
[8] Celano M, Hartmann EE, Drews-Botsch CD, Infant Aphakia Treatment Study Group Parenting stress in the infant aphakia treatment study J Pediatr Psychol: ( 2013 ) : 38: 484-493
[9] Paysse EA, Steele EA, McCreery KM Age of the emergence of negative attitudes toward strabismus J AAPOS: ( 2001 ) : 5: 361-366
[10] Uretmen O, Egrilmez S, Kose S Negative social bias against children with strabismus Acta Opthalmol Scand: ( 2003 ) : 81: 138-142
[11] Chai Y, Shao Y, Lin S Vision-related quality of life and emotional impact in children with strabismus : a prospective study J Int Med Res: ( 2009 ) : 37: 1108-1114
Coordonné par D. Denis
O. Durbec, C. Mazzeo, D. Denis
Une coopération étroite entre anesthésistes formés en pédiatrie et ophtalmopédiatres est essentielle au bon fonctionnement d'un centre d'ophtalmopédiatrie chirurgicale. L'activité est caractérisée par la succession de gestes généralement courts réalisés chez des enfants de tout âge qui sont pour la plupart pris en charge en chirurgie ambulatoire.
Nous verrons l'aspect physiologique de l'anesthésie en ophtalmologie, l'anesthésie proprement dite et les différentes pathologies rencontrées.
La connaissance des principes fondamentaux de la physiologie oculaire – pression oculaire, réflexe oculocardiaque [1] – aide à la prise en charge des enfants opérés en ophtalmologie.
La pression intra-oculaire (PIO) normale chez l'enfant varie de la naissance (10 mm) à l'adolescence (16 à 17 mmHg). Sous anesthésie, cette pression diminue de 30 à 40 % par diminution du tonus musculaire de base secondaire au sévoflurane et par déshydratation.
À l'état de base, la PIO est la résultante des pressions dues au contenu intra-oculaire sur les enveloppes du globe (plus particulièrement de la sclérotique) qui ne sont pas extensibles. Par conséquent, les variations de la PIO dépendent des variations des volumes contenus dans le globe qui sont : le volume aqueux, le volume du vitré, le volume sanguin contenu dans la choroïde. Ainsi toute augmentation de l'un de ces volumes augmente la PIO. La non-extensibilité de la sclère et du globe explique que :
- – toute pression appliquée sur le globe oculaire aboutit à une élévation de la PIO. Cette pression peut être : une manipulation chirurgicale, un hématome ou une injection intra-oculaire, un sanglage de la sclère. Une simple pression extérieure sur le globe peut entraîner une PIO> 50 mmHg;
- – tous les processus pharmacologiques ou métaboliques augmentant le volume sanguin choroïdien augmentent la congestion choroïdienne et donc la PIO.
L'anesthésiste pour maintenir une PIO normale a plusieurs moyens :
- – prévenir l'hypercapnie et l'hypoxie;
- – éviter les drogues suspectes d'augmenter la PIO : succinylcholine, kétamine;
- – réduire l'appréhension de l'enfant pour éviter les pleurs et les cris qui augmentent la pression veineuse centrale et le tonus des muscles oculomoteurs.
Le réflexe oculocardiaque (ROC) est un des témoins de l'activité vagosympathique. La voie afférente est constituée : de mécanorécepteurs périphériques orbitaires, de fibres afférentes passant par les nerfs ciliaires, du ganglion ciliaire, de la branche ophtalmique du nerf trijumeau, du ganglion de Gasser et du noyau sensitif du trijumeau. La voie efférente débute au niveau du plancher du quatrième ventricule dans le noyau du nerf vague, et atteint les structures myocardiques et nodales [2-4].
En pratique, c'est une réaction vagale consécutive à un stimulus oculaire tel qu'une traction sur les muscles oculomoteurs ou une pression externe appliquée sur le globe (compression des globes oculaires). Il se traduit par une bradycardie (baisse de plus de 10 à 30 % de la fréquence cardiaque). Ce réflexe est dÛ à une augmentation du tonus parasympatique du nerf vague.
L'arrêt de la stimulation chirurgicale est la première mesure à prendre, mais il est le plus souvent nécessaire d'administrer 10 à 20 µg/kg d'atropine ainsi que d'optimiser de l'oxygénation et de lutter contre l'hypercapnie.
Les facteurs favorisant le ROC sont essentiellement :
- – le terrain : l'enfant est un facteur favorisant, et ce d'autant qu'il présente un état anxieux;
- – la traction des muscles oculomoteurs et la pression extrinsèque exercée sur le globe oculaire.
La prise en charge péri-opératoire nécessite de disposer d'une part, d'unités d'hospitalisation pédiatriques compétentes en néonatologie avec une réanimation pédiatrique et néonatale (pour les enfants les plus graves) et d'autre part, d'un service de chirurgie ambulatoire (pour les enfants subissant des actes plus légers).
Le matériel anesthésique utilisé en pédiatrie est spécifique, ce qui explique que la mise à disposition de salles opératoires d'ophtalmologie au sein d'un « bloc pédiatrique » est la situation idéale. À défaut, l'activité d'ophtalmopédiatrie au sein d'un « bloc adulte » nécessitera beaucoup d'aménagements (circuit patient et salle de réveil spécifiques), car il est extrêmement difficile d'alterner l'activité d'adultes et d'enfants dans la même journée opératoire.
La programmation doit tenir compte du terrain. À défaut de terrains ou d'indications spécifiques, les patients les plus jeunes sont opérés en début de programme. Certains terrains peuvent être prioritaires comme les contre-indications aux gaz halogénés (myopathies, maladies mitochondriales, antécédents d'hyperthermie maligne, ec.) qui nécessitent un rinçage du circuit du respirateur.
Lors de la consultation pré-anesthésique, l'anesthésiste doit, à l'interrogatoire des parents et à l'examen clinique, rechercher systématiquement les pathologies associées les plus fréquentes en ophtalmopédiatrie : cardiaque, respiratoire ou neurologique.
L'heure du jeÛne est adaptée au programme opératoire ainsi que la convocation dans le service de chirurgie ambulatoire afin de limiter au maximum les désagréments (les anesthésies itératives pour « examen sous anesthésie générale » sont très fréquentes).
À l'issue de cette consultation, en sachant que l'hospitalisation conventionnelle est la règle chez les nourrissons avant 6 mois, la décision de réaliser le geste chirurgical prévu tient compte :
- – du niveau de compréhension de la famille;
- – de l'éloignement du domicile;
- – du terrain;
- – du geste opératoire lui-même :
-
en chirurgie ambulatoire pour : les examens ophtalmologiques, les sondages des voies lacrymales, la chirurgie des strabismes, ne nécessitant pas le plus souvent d’examen à J1 ;
en hospitalisation conventionnelle pour :
-
la surveillance ophtalmologique postopératoire immédiate des enfants de moins de 6 mois ;
les chirurgies de la cataracte et du glaucome, qui requièrent un examen à J1.
Le contrôle des voies aériennes dans la chirurgie ophtalmologique « céphalique » peut se faire au masque simple sous gaz halogéné pour un simple examen, mais le plus souvent l'utilisation d'un masque laryngé armé s'avère plus intéressante en laissant plus de place pour l'opérateur. Lors des chirurgies de la cataracte ou du glaucome, l'intubation est la technique de choix. La chirurgie du strabisme chez l'enfant plus grand peut être faite avec un masque laryngé.
La place de l'anesthésie locorégionale est limitée en ophtalmologie pédiatrique. Elle est employée en complément de l'anesthésie générale. L'anesthésie sous-ténonienne de complément est proposée dans la chirurgie du strabisme. Réalisée en début d'intervention, elle diminue l'incidence des bradycardies peropératoires ainsi que la douleur et les vomissements postopératoires.
L'antibioprophylaxie par voie générale a été remplacée par une injection intracamérulaire dans la chirurgie de la cataracte (voir sous-chapitre 5.14).
Les suites opératoires sont caractérisées par la plus grande fréquence des nausées et des vomissements (prophylaxie systématique par l'association de corticoïdes comme la dexaméthasone et d'un anti-émétique comme l'ondansétron/Zophren). La douleur postopératoire est généralement faible, excepté dans la chirurgie du strabisme. La visite anesthésique postopératoire permet de confirmer la sortie; les réorientations de l'ambulatoire vers une hospitalisation d'une nuit sont exceptionnelles.
L'existence d'une pathologie malformative ou d'un facteur de risque systémique (exemple prématurité) impose malgré un bilan ciblé une vigilance lors de chaque anesthésie générale du fait de la possible fragilité vis-à-vis de l'anesthésie [3]. De nombreuses pathologies requièrent des précautions particulières et quelques situations cliniques sont précisées ici, sans exhaustivité aucune bien entendu :
- – Prématurité. Il s'agit d'enfants nés avant la 37 semaine de grossesse pour lesquels il faut :
-
veiller à titrer l’oxygénothérapie peropératoire pour minimiser l’exposition à de hautes concentrations d’oxygène délétères ;
considérer le risque d’apnée qui est particulièrement présent jusqu’à 60 semaines d’âge post-conceptionnel ;
prévenir l’hypothermie car la perte de chaleur est très rapide chez les prématurés et nourrissons sous anesthésie générale.
- – Trisomie 21 : il faut rechercher une cardiopathie congénitale, une obstruction des voies aériennes, une instabilité du rachis cervical, une hypothyroïdie qui peut être révélée par une bradycardie jonctionnelle sous Sévorane.
- – Syndrome d'Alport : il faut rechercher une myopathie associée (éviter la succinylcholine) et/ou une insuffisance rénale associée.
- – Syndromes avec anomalies dans le développement du tissu conjonctif avec implications cardiovasculaires : syndrome de Marfan, syndrome d'Ehlers-Danlos, homocystéinurie.
- – Mucopolysaccharidoses : il s'agit de déficits enzymatiques responsables d'une dégradation incomplète de glycosaminoglycanes. La gestion des voies aériennes supérieures est généralement très difficile. L'évaluation de la fonction cardiaque est préférable dans ce contexte de pathologie infiltrative. L'accès veineux peut également s'avérer difficile.
- – Syndromes craniofaciaux : l'obstruction des voies aériennes et le rétrécissement trachéal sont la règle chez ces patients. L'intubation difficile doit être anticipée. Les craniosténoses ont une pathologie cardiaque associée.
- – Phacomatoses. Ce sont des syndromes neurocutanés avec atteintes neurologiques variables et traitements neurologiques (anti-épileptique par exemple) : taux sanguins à mesurer et bilan hépatique en péri-opératoire si besoin.
[1] Haberer JP, Obstler C Anesthésie en ophtalmologie Encycl Méd Chir: Elsevier ( 2010 ) 20- Ophtalmologie, 2010-01-01 7(4).
[2] Denis D, Wary P, Lebranchu P Physiologie oculaire Physiologie du système nerveux, Partie VI: ( 2017 ) - in press
[3] Coté CJ, Lerman J, Todres ID Practice of anesthesia in infants and children: Saunders Elsevier ( 2009 ) 1667-
[4] Bissonette B Pediatric anesthesia: Editions PMPH-USA ( 2011 ) 2255-
C. Costet, V. Desio
L'hypnose, du grec ancien Yπνoζ, dieu du sommeil, est un état de conscience modifié, entre veille et sommeil, provoqué par la suggestion. Le patient est placé dans un élément temporo-spatial différent de son quotidien, il est présent, mais indifférent à ce qui l'entoure, il ne perçoit pas d'agression. Les anesthésistes, en s'aidant de cet outil particulier qu'est l'hypnose, peuvent permettre, en milieu pédiatrique, d'améliorer la qualité et la rapidité de prise en charge de nos jeunes patients, de limiter drogues et risques opératoires, de réduire les temps périopératoires.
Il est intéressant, et même indispensable, de suivre le long chemin chaotique parcouru par l'hypnose pour comprendre son apport en médecine.
À la fin du xviii siècle, le médecin allemand Franz Messmer obtient des guérisons spectaculaires par ce qui est appelé « fluide magnétique animal » : cela marque le début, durant deux siècles, de controverses et d'une grande suspicion du milieu scientifique. En 1784, une commission d'enquête nommée par le Roi de France rejette formellement l'hypnose et l'assimile à une « médecine d'imagination » . En 1819, l'abbé Faria, ecclésiastique portugais scientifique, décrit le premier l'hypnose comme un processus naturel, dont la cause réside dans le cerveau : c'est la base de la suggestion. En 1824, le chirurgien Jules Cloquet effectue une mammectomie chez une femme pour cancer du sein sous « sommeil magnétique » seul : lui-même et sa patiente sont accusés de falsification.
Ce n'est qu'en 1890 que l'hypnose fait son entrée en neurologie, avec Jean-Martin Charcot, neurologue à la Salpêtrière : il s'agit pour lui d'un état pathologique au sein de l'hystérie (description des quatre états du « grand hypnotisme » des malades hystériques : léthargie, catalepsie, somnambulisme, amnésie). Cela devient un sujet de polémique avec les médecins Hippolyte Bernheim et Ambroise Liebaut de l'école de Nancy : pour eux, l'hypnose est un état psychologique normal, induit par la suggestion, et peut avoir des applications thérapeutiques (douleurs rhumatismales, sciatiques, aphonie, maladies de la peau, syndromes hystériques). En 1900, Sigmund Freud, après s'être rapproché de l'école de Nancy, remarque que l'hypnose peut faire apparaître ou disparaître des symptômes en rapport avec un traumatisme affectif (phénomène de « catharsis » ), mais que cela est variable selon la susceptibilité des sujets : il rejette l'hypnose pour entrer dans la psychanalyse et déclare : « Je suis en droit de dire que la psychanalyse proprement dite ne date que du jour où l'on a renoncé à avoir recours à l'hypnose. » Le milieu scientifique reste donc encore très divisé. Ce n'est qu'au milieu du xx siècle, que s'amorce un réel consensus avec la reconnaissance de différentes formes d'induction en hypnose en thérapie en 1971 par Léon Chertok et Milton Erickson. L'anesthésiste Marie-Elisabeth Faymonville met en application ces techniques et commence en 1993, dans son service grands brÛlés-chirurgie plastique, à utiliser l'hypnose en complément d'anesthésies locales et de sédatifs, en alternative à des anesthésies générales : elle rapporte, sur une série randomisée de 337 patients, une grande amélioration du confort des patients avec réduction des doses de sédatifs utilisés [1].
La reconnaissance scientifique de l'hypnose débute à partir de 1995 par le biais des équipes de P. Maquet et M.-E. Faymonville de Liège [2] et de P. Rainville de Montréal [3], en étudiant, en tomographie par émission de positrons (TEP), l'activité cérébrale par analyse des variations locales de débit sanguin cérébral (régional cerebral blood flow ou rCBF). Les comparaisons chez des patients lors de stimulations douloureuses (chaud et froid par exemple pour Rainville et al.), avant et après induction hypnotique, permettent d'objectiver des variations des rCBF (fig. 27-5 au niveau du cortex somato-pariétal (gyrus postcentral et opercule pariétal), du cortex cingulaire antérieur et des cortex occipitaux. D'autres études en électro-encéphalographie (EEG), en TEP, puis en imagerie par résonance magnétique fonctionnelle (IRMf) vont confirmer, sous hypnose, la modulation de régions corticales impliquées dans la douleur et l'activation de zones spécifiques lors de la remémoration de couleurs ou de sons [4, 5].
Fig. 27-5 Modulation de l'activité cérébrale sous hypnose.
(Source : Rainville P, Duncan GH, Bushnell MC. Représentation cérébrale de l’expérience subjective de la douleur chez l’homme. Médecine/ Sciences 2000 ; 16 : 519-27, Figure 2. http://www.ipubli.inserm.fr/bitstream/ handle/10608/1685/2000_4_519.pdf?sequence=4. Reproduction autorisée.)
L'hypnose a été largement rapportée, dans la littérature, comme bénéfique dans le domaine chirurgical adulte, notamment dans les spécialités odontologiques et obstétricales. Les auteurs, au travers d'études randomisées comparatives, retrouvent de manière quasi systématique, chez les patients opérés ayant bénéficié d'une induction en hypnose, une amélioration du confort du patient avec diminution de la douleur et de l'anxiété, moins d'hémorragies et de bronchospasmes, une utilisation moindre de sédatifs et d'antalgiques, et une récupération plus rapide [6-8]. Néanmoins deux études récentes donnent des résultats un peu discordants avec une moindre anxiété ressentie, mais pas de différence significative au niveau nausées et vomissements [9] ou au niveau administration d'antalgiques [10]. Pour l'ophtalmologie, E. Agard et al. ont récemment obtenu des résultats très positifs pour les patients, les anesthésistes, les chirurgiens, dans une étude prospective portant sur 171 patients opérés de cataracte avec préparation en hypnose [11]. La neurophysiologie en EEG, TEP, IRMf a confirmé ces dernières années, de manière objective, les modifications sous hypnose de l'activité cérébrale, avec modulation de la perception de la douleur au niveau de régions cérébrales tels les cortex cingulaires antérieurs et frontal, les ganglions de la base et le thalamus [5]. Les enfants sont plus réactifs à l'hypnose que les adultes, notamment au-delà de l'âge de 5 à 6 ans, particulièrement dans la tranche de 7 à 15 ans, mais l'induction hypnotique est difficile en deçà de l'âge de 3 ans [12]. L'hypnose est largement utilisée depuis de nombreuses années pour les douleurs chroniques des enfants, en oncologie, en stomatologie [13-18]. La littérature est, à notre connaissance, particulièrement pauvre en ophtalmologie pédiatrique [19] et en strabologie [20]. Nous avons revu les techniques d'induction en hypnose et rapporté notre expérience personnelle sur l'évolution de ces méthodes en chirurgie strabologique sur une série rétrospective de plus de 500 enfants opérés de strabismes depuis 2009. Dans les premières années, l'utilisation de métaphores, comme cela est souvent le cas en pratique de l'hypnose, était la base de notre pratique. La métaphore transporte l'enfant dans le monde imaginaire qui l'enchante (Barbie et Ken, dauphins, etc.), où la douleur est absente, où tout est beau (vidéo 27-1). Dans ces conditions, le réveil était plus tranquille, mais certains enfants restaient douloureux et agités, même si nous avions constaté une amélioration nette par rapport à nos suites opératoires antérieures sans hypnose. Cette méthode reste réservée aux enfants de moins de 3 ans ou aux enfants ayant du mal à suivre des instructions hypnotiques plus dirigistes. Pour tous les autres enfants, nous avons progressivement mis en place, comme chez les adultes, une induction dite dirigée : fixation au creux de la main, mise en contact avec les couleurs du corps (globules rouges, blancs, plaquettes jaunes, etc.), petites billes antidouleur (couleur-dilution-concentration au choix personnel), abord virtuel de la zone opérée en l'occurrence ici les yeux, coloration de cette zone avec la couleur dite morphinique (vidéo 27-2). Cette méthode permet une mise en hypnose extrêmement rapide en présence des parents, associée à la mise en place de la perfusion, injection de morphinique, d'atropine, de corticoïde et d'anti-émétique par voie parentérale. Il n'y a pas de prémédication, l'enfant passe au bloc opératoire 20 minutes plus tard (vidéo 27-3), l'intubation est très rapide. Le réveil l'est également, sans antalgique en postopératoire, le taux des enfants en réclamant est très bas, inférieur à 5 % des cas. Les anti-émétiques au réveil ne sont administrés que dans moins de 2 % des cas. Ces suites opératoires autorisent un retour au domicile rapide, avec sortie de la clinique en moyenne 3 à 5 heures après l'arrivée de l'enfant dans le service.
Vidéo 27.1 - Anesthésie pédiatrique, avec induction en hypnose par utilisation de métaphores.
Vidéo 27.2 - Anesthésie pédiatrique, avec induction en hypnose dite dirigée.
Vidéo 27.3 - Arrivée d’un enfant au bloc opératoire, après induction en hypnose, sans prémédication préalable.
En ophtalmopédiatrie chirurgicale et en strabologie, l'hypnose pourrait ainsi permettre :
- – en préopératoire : d'éviter une prémédication, d'obtenir un gain de temps sous réserve d'une équipe médicale bien rodée à la méthode;
- – en peropératoire : d'obtenir un meilleur équilibre hémodynamique, un taux moindre de bronchospasmes, une extubation plus rapide;
- – en postopératoire : un réveil calme, avec un taux d'administration d'antalgiques nettement réduit, une hospitalisation d'autant raccourcie.
Ces données demandent encore à être validées dans notre spécialité par des études prospectives randomisées au long cours, comme cela a déjà été le cas dans d'autres spécialités pédiatriques, notamment dans les unités médico-chirurgicales où les enfants sont particulièrement confrontés au problème de la douleur [13, 14, 17].
Après de nombreuses années de controverses, l'hypnose a acquis, depuis sa validation par les neurosciences, sa place dans le domaine médico-chirurgical. Les communications dans la littérature se multiplient ces dernières années dans le domaine pédiatrique, notamment dans les spécialités où les enfants sont confrontés au problème de la douleur (oncologie, grands brÛlés). En ophtalmopédiatrie et strabologie, cette pratique reste encore très confidentielle, même si elle paraît apporter un gain non négligeable de confort, de qualité de soins, pour toute l'unité enfants-parents et équipe médicale anesthésie-chirurgie. Les résultats positifs déjà rapportés encouragent à la mise en place d'études randomisées et au développement de formations spécifiques des équipes de bloc opératoire. Dans notre pratique chirurgicale, l'hypnose permet le raccourcissement de la durée d'hospitalisation, par amélioration des conforts de soins.
[1] Faymonville ME, Fissette J, Mambourg PH Hypnosis as adjunct therapy in conscious sedation for plastic surgery Reg Anesth: ( 1995 ) : 20: 145-151
[2] Maquet P, Faymonville ME, Degueldre C Functional neuroanatomy of hypnotic state Biol Psychiatry: ( 1999 ) : 45: 327-333
[3] Rainville P, Duncan GH, Price DD Pain affect encoded in human anterior cingulate but not somatosensory cortex Science: ( 1997 ) : 277: 968-971
[4] Apkarian VA, Bushnell MC, Treede RD, Zubieta JK Human brain mechanisms of pain perception and regulation in health and disease Eur J Pain: ( 2005 ) : 9: 463-484
[5] Vanhaudenhuyse A, Laureys S, Faymonville ME Neurophysiology of hypnosis Neurophysiol Clin: ( 2014 ) : 44: 343-353
[6] Enqvist B, Björklund C, Engman M, Jakobsson J Preoperative hypnosis reduces postoperative vomiting after surgery of the breasts. A prospective, randomized and blinded study Acta Anaesthesiol Scand: ( 1997 ) : 41: 1028-1032
[7] Musellec H, Bernard F, Houssel P Ambulatory essure implant placement sterilization procedure for women : prospective study comparing general anesthesia versus hypnosis combined with sedation Ann Fr Anesth Reanim: ( 2010 ) : 29: 889-896
[8] Abdeshahi SK, Hashemipour MA, Mesgarzadeh V Effect of hypnosis on induction of local anaesthesia, pain perception, control of haemorrhage and anxiety during extraction of third molars : a case-control study J Craniomaxillofac Surg: ( 2013 ) : 41: 310-315
[9] Izanloo A, Fathi M, Izanloo S Efficacy of conversational hypnosis and propofol in reducing adverse effects of endoscopy Anesth Pain Med: ( 2015 ) : 5: e27695-
[10] Cyna AM, Crowther CA, Robinson JS Hypnosis antenatal training for childbirth : a randomised controlled trial BJOG: ( 2013 ) : 120: 1248-1259 discussion 1256-7
[11] Agard E, Pernod C, El Chehab H A role for hypnosis in cataract surgery : report of 171 procedures J Fr Ophtalmol: ( 2016 ) : 39: 287-291
[12] Rogovik AL, Goldman RD Hypnosis for treatment of pain in children Can Fam Physician: ( 2007 ) : 53: 823-825
[13] Kuttner L, Bowman M, Teasdale M Psychological treatment of distress, pain, and anxiety for young children with cancer J Dev Behav Pediatr: ( 1988 ) : 9: 374-381
[14] Wild MR, Espie CA The efficacy of hypnosis in the reduction of procedural pain and distress in pediatric oncology : a systematic review J Dev Behav Pediatr: ( 2004 ) : 25: 207-213
[15] Calipel S, Lucas-Polomeni MM, Wodey E, Ecoffey C Premedication in children : hypnosis versus midazolam Paediatr Anaesth: ( 2005 ) : 15: 275-281
[16] Huet A, Lucas-Polomeni MM, Robert JC Hypnosis and dental anesthesia in children : a prospective controlled study Int J Clin Exp Hypn: ( 2011 ) : 59: 424-440
[17] Tomé-Pires C, Miró J Hypnosis for the management of chronic and cancer procedure-related pain in children Int J Clin Exp Hypn: ( 2012 ) : 60: 432-457
[18] Adinolfi B, Gava N Controlled outcome studies of child clinical hypnosis Acta Biomed: ( 2013 ) : 84: 94-97
[19] Lewenstein LN Hypnosis as an anesthetic in pediatric ophthalmology Anesthesiology: ( 1978 ) : 49: 144-145
[20] Medková L, Tejklová M Use of hypnosis in certain forms of childhood strabismus Cesk Oftalmol: ( 1992 ) : 48: 374-376
- Chapitre 27
Dialogue entre ophtalmologistes et pédiatres – interdisciplinarité- 1 – Introduction : de l’interdisciplinarité en ophtalmologie pédiatrique
- 2 – Dialogue entre infectiologue et ophtalmologiste
- 3 – Dialogue entre rhumatologue et ophtalmologiste
- 4 – Dialogue entre neuropédiatre et ophtalmologiste
- 5 – Dialogue entre dermatologue et ophtalmologiste
- 6 – Dialogue entre oncologue et ophtalmologiste
- 7 – Dialogue entre hématologue et ophtalmologiste
- 8 – Dialogue entre endocrinologue et ophtalmologiste
- 9 – Dialogue entre hépato-gastro-entérologue et ophtalmologiste
- 10 – Dialogue entre cardiologue et ophtalmologiste
- 11 – Dialogue entre pneumologue et ophtalmologiste
- 12 – Dialogue entre néphrologue et ophtalmologiste
- 13 – Dialogue entre oto-rhino-laryngologiste pédiatre et ophtalmologiste
- 14 – Dialogue entre chirurgien maxillofacial et ophtalmologiste
- 15 – Dialogue entre neurochirurgien et ophtalmologiste : que faire si mon enfant a une déformation crânienne ?
- 16 – Dialogue entre psychiatre et ophtalmologiste
- 17 – Dialogue entre anesthésiste et ophtalmologiste
- Anesthésie pédiatrique et ophtalmologie
- Apport de l’hypnose en anesthésie pédiatrique