Pathologie des paupières et des voies lacrymales
Coordonné par D. Denis
O. Galatoire
Les anomalies des paupières de l’enfant sont une source d’angoisse pour les parents. La modification de l’aspect du regard « idéal » est un motif de préoccupation avec un souhait de « normalité » sous-jacent. Les anomalies palpébrales peuvent être isolées, rentrer dans le cadre de syndromes malformatifs plus complexes avec des modifications locales intéressant à la fois la face, l’orbite et le globe oculaire.
Dans certains cas, ces anomalies s’intègrent dans le cadre de pathologies plus générales, d’un syndrome polymalformatif nécessitant une prise en charge multidisciplinaire de l’enfant.
La prise en charge reposera tout d’abord sur un bilan ophtalmologique avec évaluation de la statique et dynamique palpébrale.
La recherche de manifestations systémiques réalisée en collaboration avec le pédiatre sera systématique. Un bilan d’imagerie est parfois nécessaire, il reposera sur la réalisation d’une échographie orbitaire, avec scanner et reconstruction osseuse dans certains cas, et enfin d’une imagerie par résonance magnétique (IRM) pour les syndromes les plus complexes.
La prise en charge médico-chirurgicale dépendra du type d’anomalie et de l’âge de l’enfant.
Pendant la prime enfance, le but sera d’assurer la protection du globe oculaire tout en permettant un développement satisfaisant à la fonction visuelle. Pour les enfants plus grands, les considérations esthétiques et la restauration de l’apparence normale seront considérées.
Nous distinguerons les malformations palpébrales qui correspondent à une altération de la structure même de la paupière et les malpositions palpébrales qui correspondent à une anomalie de positionnement statique ou dynamique d’une paupière normalement formée (Fig. 7-1).
Des anomalies malformatives exceptionnelles type ablépharie ou cryptophtalmie ne seront pas traitées dans ce chapitre.
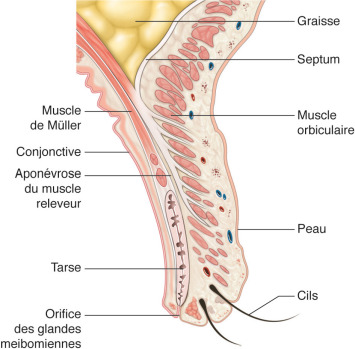
Fig. 7-1 Coupe de paupières représentant les différents tissus.
L’épicanthus constitue un repli cutané qui part de la racine du nez et se projette en regard de la commissure interne. Il est presque toujours bilatéral, mais le plus souvent asymétrique.
Lors de la croissance de l’enfant, le visage s’allonge de façon antéropostérieure et l’épicanthus disparaît. Il subsiste néanmoins chez 2 à 5 % des Caucasiens et 70 à 90 % des Asiatiques, chez qui il peut être considéré comme physiologique. On distingue différents types d’épicanthus en fonction de l’aspect du pli cutané par rapport au bord libre palpébral (eFig. 7-1) :
épicanthus supracilliaris : débute au niveau de la région sousciliaire, traverse le canthus interne en regard du sac lacrymal pour rejoindre la paupière inférieure;
épicanthus palpebralis : débute au niveau de la paupière supérieure, au-dessus du tarse et va s’étendre jusqu’au niveau du rebord orbitaire inférieur;
épicanthus tarsalis : il naît dans le pli palpébral pour rejoindre le canthus interne;
épicanthus inversus : débute en paupière inférieure pour remonter au niveau de la paupière supérieure qui est le plus souvent préservée.
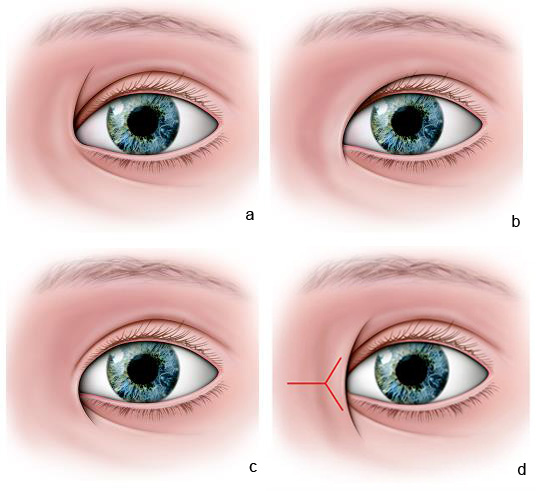
eFig. 7-1 Les différents types d’épicanthus
a. Épicanthus supraciliaris, b. Épicanthus tarsalis, c. Épicanthus palpebralis, d. Épicanthus inversus
L’épicanthus inversus, qui ne régresse pas avec l’âge, nécessite le plus souvent une correction chirurgicale. Il est souvent associé à un télécanthus, notamment dans le cadre d’un syndrome de blépharophimosis. On privilégiera une prise en charge à l’issue de la croissance faciale. Les autres épicanthus (supracillaris, palpébralis et tarsalis) sont souvent peu marqués et ont tendance à régresser spontanément [1].
Le télécanthus interne correspond à une augmentation de distance entre les deux canthi internes, alors que les distances interpupillaires et intercanthales externes sont normales. Ainsi, la sclère en interne du limbe n’est plus visible. On distinguera le télécanthus de l’hypertélorisme qui correspond à une augmentation de l’espace interorbitaire et qui se traduit par une augmentation de la distance entre les globes oculaires. Pour distinguer les deux affections cliniques, on mesure la distance intercanthale interne, la distance intercanthale externe et la distance interpupillaire. Une prise en charge chirurgicale est le plus souvent nécessaire. Le télécanthus du canthus latéral est exceptionnel, il se rencontre le plus souvent dans le cadre d’euryblépharon, du syndrome de Waardenburg ou de dystopie médiocanthale. Dans ce syndrome malformatif, le télécanthus est bilatéral isolé, sans épicanthus associé [2].
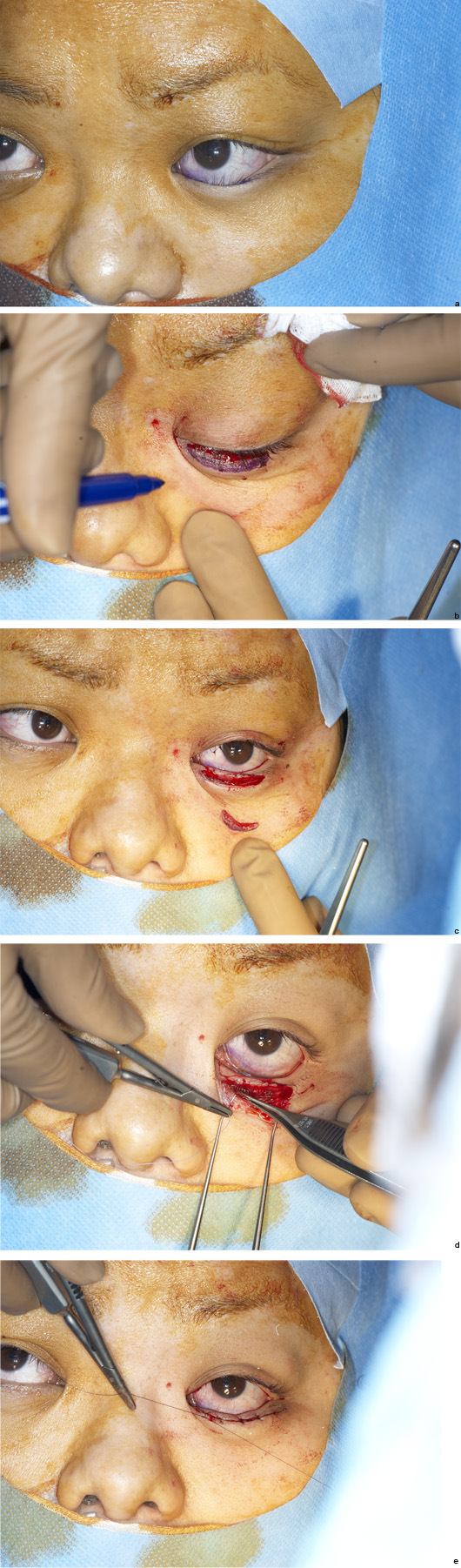
La majorité des patients bénéficient d’une plicature du tendon canthomédial. Cette technique est suffisante lors de déplacement latéral modéré du canthus médial. Ce geste chirurgical permet d’éviter la canthopexie transnasale plus invasive. Différentes plasties cutanées peuvent être associées à la plicature du tendon canthal médial. La technique Y-V peut être réalisée lors d’absence d’épicanthus associé. Lorsqu’il existe un repli épicanthal marqué, notamment lors des syndromes de blépharophimosis, nous préférons réaliser une plastie de Mustardé (Fig. 7-2 et eFig. 7-2) ou encore une plastie d’Anderson et Novinski [3].
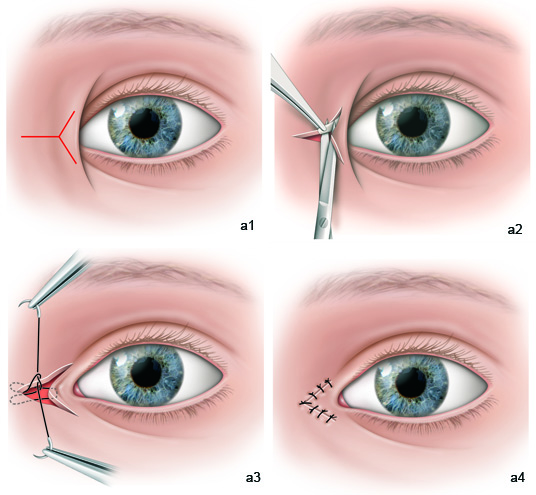
eFig. 7-2 Cure de l’épicanthus et du télécanthus par double plastie en Z de Mustardé (a) et plastie Y-V (b)
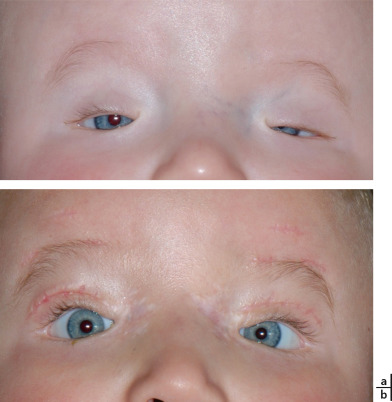
Fig. 7-2 Syndrome de blépharophimosis (a).
Résultat précoce après canthoplastie interne selon Mustardé et suspension frontale bilatérale (b).
Cette intervention chirurgicale est nécessaire lors de télécanthi majeurs. Elle sera réalisée à l’âge de 4 ou 5 ans, lorsque le massif facial osseux aura atteint une certaine maturité. La réalisation d’un scanner orbitaire est nécessaire afin de mettre en évidence la lame papyracée qui peut être située en position plus basse lors de malformations associées.
Si le télécanthus est associé à un épicanthus, il faudra réaliser également une plastie cutanée (eFig. 7-3) [4].
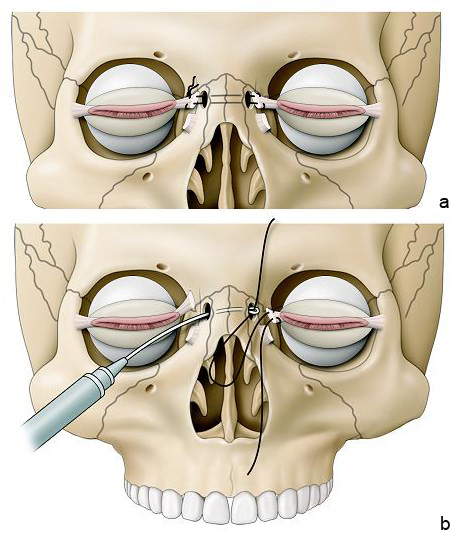
eFig. 7-3 Canthopexie transnasale
Ces anomalies constitutionnelles consistent en une antéposition du chef antérieur du tendon canthal interne. Cette anomalie n’est pas gênante, parfois non détectée dans la prime enfance du fait de la base du nez qui est encore plate. Lorsque l’enfant grandit, notamment à l’adolescence, le signe fonctionnel le plus fréquent est un larmoiement clair. La position antérieure du tendon canthal médial entraîne un diastasis oculopalpébral avec un écartement entre le point lacrymal et le globe oculaire. Le traitement est chirurgical. Il consiste à réaliser une plastie cutanée en V-Y [5].
La malposition canthale externe correspond à une anomalie de l’obliquité de la fente palpébrale, souvent associée à des anomalies osseuses sous-jacentes. Chez l’enfant caucasien normal, la fente palpébrale est légèrement oblique en haut et en dehors avec un canthus latéral plus haut de 1 à 2 mm que le canthus médial.
Cette anomalie entraîne une obliquité mongoloïde de la fente palpébrale.
Le canthus latéral est plus haut situé que le canthus médial avec une fente palpébrale orientée en haut et en dehors.
Hormis l’aspect retrouvé chez les Asiatiques, certains syndromes dysmorphiques, tels la trisomie 21 ou syndrome de Down, présentent une dystopie supérieure. Elle est associée dans ces cas précis à un retard mental, des anomalies plus générales diverses. La trisomie 21 est appelée mongolisme du fait de l’accentuation de la fente palpébrale vers le haut comparable à l’aspect que présentent certaines populations asiatiques. Cette dysmorphie associe une brachycéphalie, une face ronde et des lèvres épaisses. Le plus souvent, il n’y a pas d’indication à un traitement chirurgical [6].
Dans ce cas, la fente palpébrale est orientée vers le bas avec une obliquité dite « antimongoloïde » de la fente palpébrale, le canthus latéral est plus bas que le canthus médial. On le retrouve dans le syndrome de Franceschetti ou encore dans le chromosome 21 en anneau ou dans le contretype de la trisomie 21.
Le syndrome de Franceschetti, ou dysostose mandibulofaciale, décrit en 1949 par Franceschetti, correspond à une dysostose mandibulofaciale associée à une dysmorphie caractéristique. Outre l’obliquité antimongoloïde bilatérale et bipalpébrale, les paupières inférieures présentent le plus souvent un colobome de la partie latérale. Des anomalies du massif osseux facial sont associées avec une hypoplasie de l’os malaire, des mandibules, la présence des fentes faciales 6, 7, 8 de la classification de Tessier. Une malformation de l’oreille externe, voire de l’oreille moyenne et interne, est retrouvée dans certains cas [7].
Il a été distingué en 1968 par Noonan du syndrome de mâle de Turner. Cette anomalie regroupe une dysmorphie avec hypertélorisme, une obliquité antimongoloïde et fente palpébrale, un épicanthus, un ptosis et un pterygium colli. Les atteintes oculaires associées sont les strabismes, les cataractes, une dystrophie cornéenne stromale antérieure, les hypoplasies du nerf optique et les colobomes choriorétiniens. Au point de vue général, on peut retrouver des anomalies cardiaques, squelettiques, une petite taille, un pectus carinatus ou excavatus, un cou palmé, une anomalie génitale (cryptorchidie), une anomalie sanguine (trouble de la coagulation) ou encore une surdité. Le retard intellectuel est présent dans 4 % des cas.
En l’absence d’ectropion associé avec bonne couverture oculaire, le traitement chirurgical n’est pas nécessaire chez le petit enfant. Lors de la croissance et l’apparition de considérations plastique et/ou esthétique, le traitement consiste en un repositionnement du canthus latéral par une canthopexie externe. Pour les syndromes de Franceschetti avec notamment un colobome latéral, une chirurgie plus complexe d’allongement de paupières par greffe ou lambeau est parfois nécessaire. La méthode la plus appropriée consiste à utiliser un lambeau à charnière latérale permettant ainsi de traiter la brièveté palpébrale latérale (Fig. 7-3).
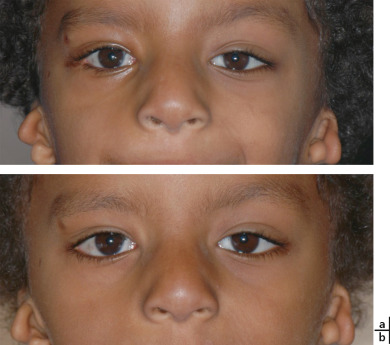
Fig. 7-3 Syndrome de Franceschetti.
a. Malformation canthus externe et oreille. b. Résultat postopératoire après plastie du canthus externe.
Le ptosis correspond à une réduction de la fente palpébrale dans son axe vertical par la ptose de la paupière supérieure.
Il est consécutif à la diminution d’action de deux muscles releveurs de la paupière supérieure : le muscle de la paupière supérieure innervé par la 3e paire crânienne et/ou le muscle de Müller innervé par le système sympathique.
C’est l’étiologie la plus fréquente des ptosis congénitaux. On parle souvent de ptosis congénital simple. Le ptosis myogène est consécutif à une anomalie du développement embryonnaire des fibres du muscle releveur de la paupière supérieure. Le muscle est d’aspect dystrophique, fibreux et sa fonction en est réduite. L’élévation de la paupière supérieure est mauvaise et parfois très insuffisante. L’élasticité musculaire est également insuffisante entraînant une absence de déroulé de la paupière lors du regard vers le bas avec parfois une lagophtalmie au cours du sommeil. Dans certains cas, ce ptosis myogène est associé à une anomalie de l’action du muscle droit supérieur. Le ptosis myogène peut également s’intégrer dans un syndrome polymalformatif modéré, voire complexe, comme dans certaines dysmorphies ou encore le syndrome de blépharophimosis.
Dans ce cas, les fibres musculaires ne sont pas atteintes, mais il s’agit d’une anomalie de la commande nerveuse par trouble du développement innervationnel durant l’embryogenèse ou encore par atteinte nerveuse secondaire (traumatisme, tumeur, inflammation). On distingue l’atteinte du nerf oculomoteur (3e paire crânienne) qui dans sa forme complète, extrinsèque et intrinsèque associera un ptosis, une mydriase aréactive et une paralysie de l’élévation et de l’adduction du globe, ce dernier étant dévié en bas et en dehors. La plupart des atteintes du nerf oculomoteur sont partielles avec des ptosis et une déviation oculaire variables.
Le syndrome de Claude-Bernard-Horner congénital associe un ptosis, un myosis, une énophtalmie homolatérale (ou pseudo-énophtalmie) avec dépigmentation irienne. Il est la conséquence d’une atteinte du plexus sympathique, le plus souvent la ptose palpébrale est modérée. La prise en charge est fonction de l’importance du ptosis.
Le syndrome de Marcus-Gunn correspond à des connexions anormales entre le nerf III et le contingent moteur du nerf V, responsables de syncinésies oropalpébrales. Les mouvements de la mandibule, notamment la mastication ou encore la déglutition, provoque une élévation d’une paupière ptosée donnant l’impression que l’enfant cligne de l’œil. Cet aspect est constaté dès les premiers mois de vie par les parents.
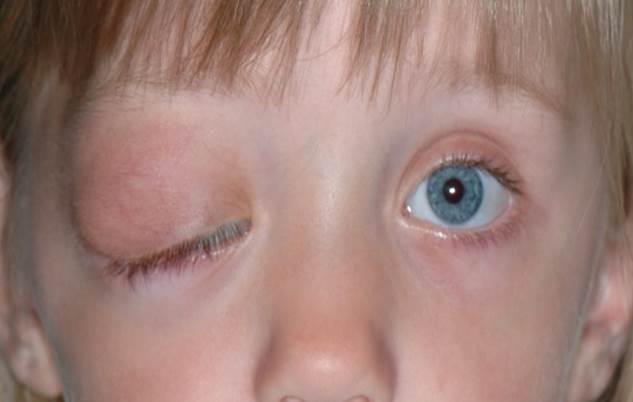
Le ptosis mécanique de l’enfant est essentiellement dÛ au développement d’une masse tumorale palpébrale. L’hémangiome capillaire infantile de l’enfant reste l’étiologie la plus fréquente. Si l’exérèse chirurgicale était la règle, lors de ptosis mécanique couvrant l’axe visuel ou menaçant le développement fonctionnel, la prise en charge a été révolutionnée par la prise en charge médicale et notamment les bêtabloquants par voie orale. Cette classe thérapeutique permet une régression importante de cet hémangiome bénin, reléguant l’exérèse chirurgicale à de rares cas résistants.
La neurofibromatose de type I avec la présence de névromes plexiformes venant alourdir la paupière supérieure est également une étiologie connue de ptosis mécanique de l’enfant. Un épaississement infiltratif de l’ensemble de la paupière supérieure peut également être associé. La prise en charge consiste alors à réaliser une ablation des névromes plexiformes et un amincissement des tissus cutanés et sous-cutanés.
Le traitement passe dans un premier temps par la levée de la compression.
C. Beaube-Bok
Le risque d’amblyopie doit être évalué dans la prise en charge initiale d’un ptosis. Expérimentalement, l’occlusion palpébrale complète unilatérale conduit en quelques jours à une amblyopie dite de déprivation (il serait plus juste de dire privation) de l’œil considéré que ce soit chez l’être humain ou d’autres espèces animales. Le diagnostic d’amblyopie de privation, donc liée uniquement au ptosis, est posé dès lors que l’amblyopie ne peut être reliée à aucun autre facteur amblyogène tel qu’une erreur réfractive, un strabisme ou une anomalie oculaire organique [8].
Dans notre pratique, les formes isolées de ptosis représentent la grande majorité des cas (70 % ). Dans ces formes, l’importance moyenne du ptosis est la plus fréquente (75 % ). Un astigmatisme direct compris entre 2 et 4 D est l’amétropie la plus fréquemment rencontrée, mesurée dans 67 % de nos cas. La fréquence de l’amblyopie de privation dans nos cas de ptosis isolés est de 2,6 % [9], rejoignant les données de la littérature [10 - 12]. Le ptosis isolé est peu amblyogène.
Les formes associées à des troubles oculomoteurs présentent un fort risque d’amblyopie liée au strabisme et à l’anisométropie [13 - 15]. Dans les ptosis syndromiques, les paralysies du III et les ptosis mécaniques, l’amblyopie est retrouvée dans 93 à 100 % de nos cas. Ces formes associent au ptosis des troubles oculomoteurs, une amétropie et/ou des anomalies organiques importantes. L’amblyopie est donc dans ces formes de causes multiples.
En résumé, l’amblyopie dans les ptosis (syndromique ou non) est le plus souvent multifactorielle. Le risque d’amblyopie est d’autant plus grand que le ptosis est unilatéral majeur, associé à des troubles oculomoteurs et/ou une anisométropie, ou syndromique. L’unilatéralité est un facteur de risque plus important que l’importance du ptosis [16 - 18]. Il est essentiel, lors de la prise en charge initiale, de faire la part des choses entre les différents facteurs amblyogènes, afin de traiter en priorité l’amblyopie par correction optique adaptée ± occlusion. L’indication chirurgicale précoce ne doit être posée que si le diagnostic d’amblyopie par privation est retenu ou s’il existe un torticolis invalidant.
La plupart des ptosis congénitaux sont des ptosis myogènes avec mauvaise fonction du muscle releveur. La chirurgie du muscle releveur ne permettra pas de rendre une fonction normale au releveur de la paupière. Le but de l’intervention chirurgicale est d’obtenir une ouverture de la fente palpébrale, la plus symétrique possible en position primaire. Lors du regard vers le haut, la paupière remontera moins que celle controlatérale et dans le regard vers le bas, elle descendra moins également. Il est indispensable d’en prévenir les parents. Par ailleurs, en cas de faible fonction du muscle releveur de la paupière supérieure, le traitement chirurgical, même s’il est parfaitement mené, ne permettra pas toujours une stabilité du résultat dans le temps. Pour des raisons mécaniques, il y aura une régression du résultat au fur et à mesure de la croissance de l’enfant. La résection maximale du muscle releveur ou encore la suspension frontale, système statique, est contrebalancée par un système dynamique dont la fonction est bonne, à savoir la fermeture palpébrale par la contraction d’un muscle orbiculaire puissant et fonctionnel. Ainsi le montage chirurgical pour lever la paupière supérieure finira par se distendre avec une récidive plus ou moins importante de la ptose palpébrale nécessitant parfois une réintervention. Lors de ptosis congénital isolé bilatéral, la symétrisation est plus facile et on hésitera moins à réaliser une suspension frontale bilatérale. En effet, dans ces cas-là, on ne sera pas confronté à une asymétrie avec une suspension frontale d’un côté et un muscle fonctionnel de l’autre [19].
La stratégie chirurgicale sera à définir en fonction des cas et après discussion avec l’entourage et les parents. Une des difficultés réside dans le fait que le visage de l’enfant va grandir et les tissus palpébraux vont se développer. Pour le nourrisson, la prévention de l’amblyopie est l’élément le plus important à prendre en considération pour l’indication chirurgicale [20].
Lors d’une obstruction de l’axe visuel complète, l’intervention chirurgicale sera à réaliser précocement à savoir à 2-3 mois de vie, dans le but de dégager l’axe visuel. Les parents seront avertis que des réinterventions à titre cosmétique seront nécessaires du fait de la croissance de l’enfant. Si la fonction du muscle releveur de la paupière supérieure est très faible ou nulle, la suspension de la paupière au muscle frontal sera la meilleure technique, permettant de dégager l’axe visuel. En effet, le raccourcissement du muscle releveur de la paupière supérieure dans ces cas-là ne donnera pas un résultat satisfaisant et ne préviendra pas l’amblyopie. Pour cela, chez le nourrisson chez qui le prélèvement de matériel autologue (fascia lata) est difficile, nous privilégierons la suspension par bande de polytétrafluoroéthylène (PTFE) ou encore par un tube de silicone. En l’absence de menace sur le développement visuel, l’intervention pourra être réalisée entre 3 et 5 ans [21].
Opérer ces enfants avant l’âge de 3 ans ne permettra pas toujours de réaliser un geste durable du fait du faible développement des tissus palpébraux. Par ailleurs, il paraît souhaitable de traiter l’enfant avant 5 ans (avant la rentrée en cours primaire), afin de lui éviter les éventuels troubles de socialisation qui pourraient en découler. L’intervention se déroule sous anesthésie générale, l’adaptation du geste chirurgical en évaluant pendant l’intervention la fonction du muscle releveur n’est pas possible. La technique la plus couramment utilisée est celle d’un raccourcissement du muscle releveur. La discussion et l’exposition des principes du ptosis et de sa prise en charge sont indispensables auprès des parents.
Le choix de la technique chirurgicale variera en fonction de l’étiologie du ptosis et de la fonction du muscle releveur de la paupière supérieure (Tableau 7-1).
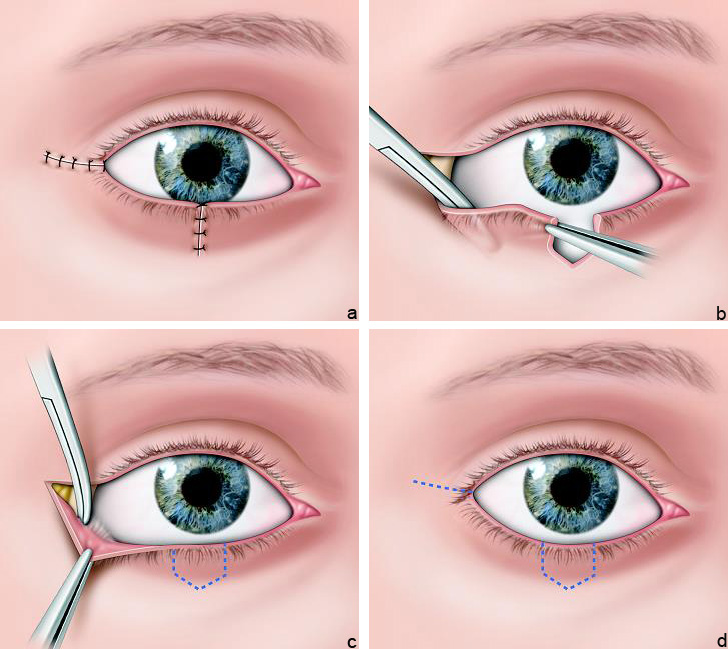
Il s’agit de la technique la plus souvent utilisée pour la prise en charge du ptosis congénital. La voie d’abord utilisée est plus souvent antérieure cutanée ou plus rarement conjonctivale. Le principe consiste à raccourcir le muscle releveur pour soulever la paupière.
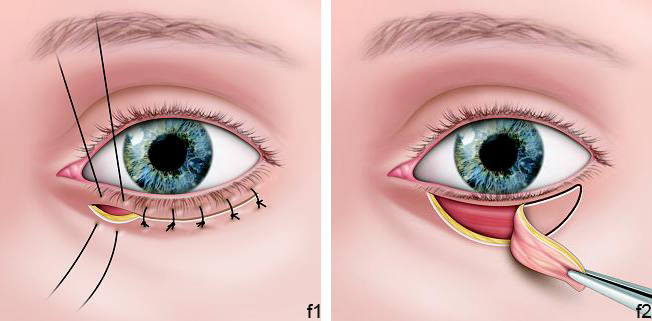
La voie antérieure présente plusieurs avantages (eFig. 7-4). Elle permet tout d’abord de réaliser une éventuelle résection cutanée et orbiculaire, d’exposer parfaitement le muscle releveur et de pouvoir ainsi réaliser une résection maximale de manière beaucoup plus aisée que par voie conjonctivale. La constatation peropératoire d’un muscle de trop mauvaise qualité pourra orienter le praticien vers une modification de l’indication et l’amènera alors à réaliser une suspension de la paupière au muscle frontal. La voie d’abord antérieure permet en outre de maîtriser parfaitement la position du pli en procédant à des sutures en fin d’intervention du plan superficiel au plan profond qui vont provoquer une adhérence et créer ainsi un pli [22].
Le niveau de résection musculaire réalisé sur le muscle releveur varie en fonction de l’importance du ptosis et de la fonction du muscle releveur (Tableau 7-2 et 7-3).
De nombreux auteurs ont établi des règles chirurgicales indiquant l’importance de la résection musculaire réalisée en fonction de l’importance du ptosis. Nous distinguerons trois ensembles de fonction du releveur : faible < 4 mm; moyenne de 5 à 7 mm; bonne > 8 mm. Une fonction faible ou absente conduira à réaliser une suspension frontale. Une fonction moyenne et satisfaisante conduira à une résection musculaire (Fig. 7-4).
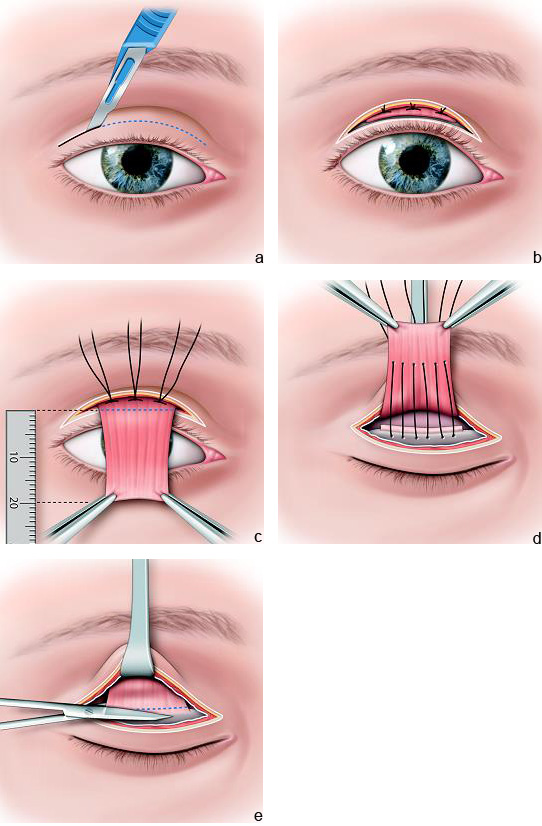
eFig. 7-4 Raccourcissement du releveur par voie antérieure
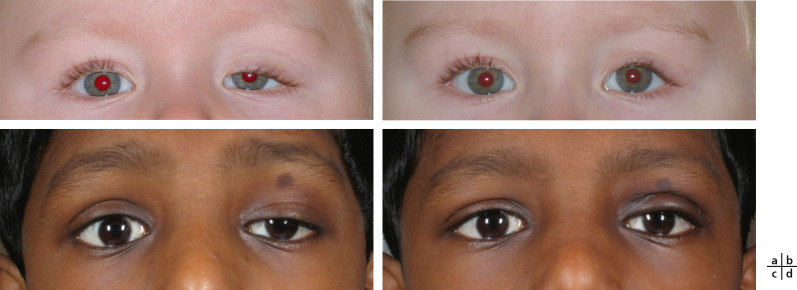
Fig. 7-4 Ptosis congénital unilatéral opéré selon la technique de la résection du muscle releveur (a-d) avec reformation du pli palpébral (c, d).
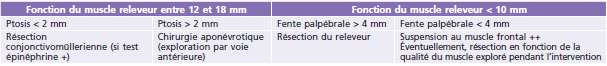
Tableau 7-1 Choix d’une technique chirurgicale d’un ptosis isolé selon la fonction du muscle releveur de la paupière supérieure.

Tableau 7-2 Dosage de la résection du releveur.
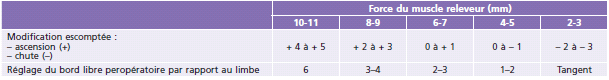
Tableau 7-3 Réglage peropératoire de la hauteur du bord libre.
Cette technique est utilisée pour les ptosis les plus importants avec une faible fonction du muscle releveur. Lors de la suspension de la paupière au muscle frontal, la fermeture devient active par contraction du muscle orbiculaire. L’ouverture est passive et majorée par la contraction du muscle frontal. Cette technique permet de suppléer l’insuffisance du muscle releveur par le muscle frontal. La contraction du muscle orbiculaire associée à un relâchement de la contraction du muscle frontal permettra d’abaisser le sourcil et ainsi la paupière.
Cette technique est préférable à une résection trop importante du muscle releveur qui peut fixer la paupière et rendre son occlusion plus difficile par l’absence complète de souplesse du muscle releveur. Un des inconvénients en revanche de la suspension frontale est celui de l’asynergie oculopalpébrale, à savoir lorsque le patient regarde vers le bas, la paupière supérieure perd son déroulé. Cet aspect est d’autant plus gênant qu’il s’agit d’une suspension unilatérale. Ainsi, certains auteurs préconisent de réaliser une suspension frontale bilatérale pour la prise en charge des ptosis majeurs unilatéraux. Cette stratégie thérapeutique est largement discutée, elle peut également être utilisée dans un deuxième temps en cas d’inconfort ou d’aspect inesthétique dÛ à cette asynergie (Fig. 7-5).
Cette technique est rarement utilisée pour la prise en charge des ptosis congénitaux. Cette technique consiste à réaliser un raccourcissement du muscle de Müller à la face postérieure de l’aponévrose du muscle releveur de la paupière (eFig. 7-5).
Elle est indiquée lors de positivité du test à l’épinéphrine. Celui-ci consiste à instiller une goutte d’épinéphrine du côté de la paupière ptosée. Ce test est positif si on constate que la paupière ptosée remonte. Cette technique est utilisée essentiellement pour les syndromes de Claude-Bernard-Horner congénitaux ou périnatals qui sont rares.
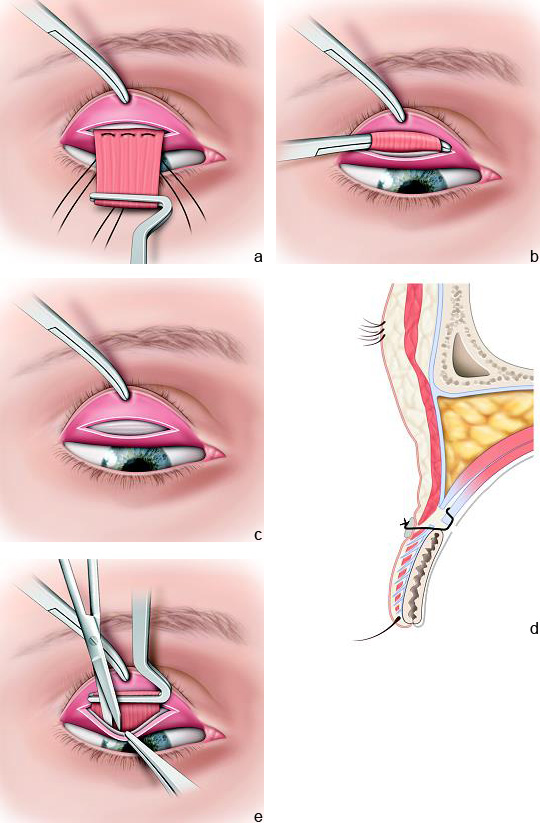
eFig. 7-5 Raccourcissement du releveur par voie conjonctivale
Cette technique consiste à réaliser une plicature, voire un raccourcissement limité au niveau de l’aponévrose du muscle releveur. L’abord chirurgical est identique à celui réalisé pour le raccourcissement musculaire. Cette technique diffère par le fait que l’on n’effectue pas de résection musculaire, mais simplement un rapprochement, voire une plicature de l’aponévrose du releveur. Cette technique est susceptible d’entraîner des surcorrections postopératoires du fait de l’absence de dosage possible et de l’anesthésie générale, lors de la prise en charge de ces ptosis modérés.
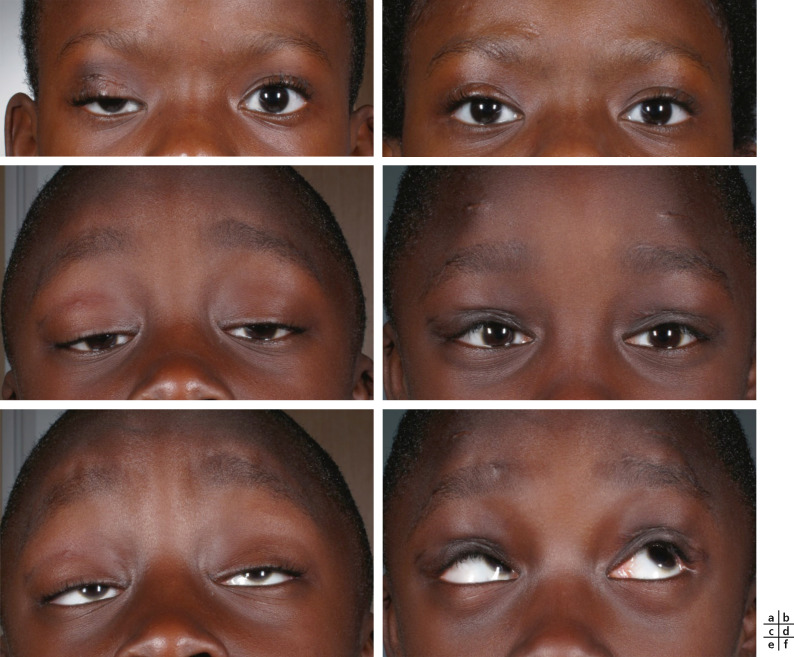
Fig. 7-5 Ptosis unilatéral traité par une suspension frontale unilatérale selon la technique de Fox modifiée par Morax.
a, c, e. Préopératoire. b, d, f. Résultat en élévation permettant de retrouver un champ visuel supérieur.
Lors de malformations orbitopalpébrales complexes, la prise en charge devra débuter par la correction des anomalies osseuses si elle est nécessaire, puis l’œil devra être recentré par une chirurgie oculomotrice et enfin l’ajustement palpébral sera réalisé. Cet ordre dans la prise en charge est nécessaire à l’obtention d’un résultat satisfaisant. La collaboration entre les différents intervenants est souhaitable.
Lors d’une paralysie de la 3e paire crânienne, le ptosis est associé à des anomalies oculomotrices; le globe est dévié en dehors et en bas. La prise en charge de ce ptosis est particulièrement complexe.
Le premier temps consistera en une chirurgie oculomotrice, permettant de recentrer l’œil en position primaire. Si du fait de l’absence de commande nerveuse, la fonction du muscle releveur n’est pas exploitable, une suspension de la paupière au muscle frontal sera lors indiquée. Celle-ci devra être particulièrement prudente, l’absence de réflexe de Charles Bell, avec troubles de l’élévation de l’œil, expose tout particulièrement l’enfant à un risque de lagophtalmie et d’exposition cornéenne. La suspension frontale sera néanmoins préférée à la résection maximale du releveur, du fait de la souplesse par fonctionnalité du muscle frontal [23].
La prise en charge du syndrome de Marcus-Gunn est complexe et le rapport risques/bénéfices sera bien pesé avant de prendre une décision thérapeutique. La prise en charge est variable en fonction de l’importance du ptosis. En cas de ptosis discret et de gêne fonctionnelle minime, l’abstention thérapeutique est la règle. En cas de ptosis important avec syncinésie handicapante, une intervention lourde avec dénervation du muscle releveur et suspension frontale est alors nécessaire. Elle seule permet la suppression de l’asynergie oculopalpébrale. Cette intervention sera réalisée à l’adolescence. En effet, les régressions spontanées à évolution favorable peuvent émailler les premières années de vie.
Celle-ci est diagnostiquée lorsque la paupière supérieure est située au-dessus du limbe. Elle est le plus souvent asymétrique, due à une anomalie du muscle releveur et/ou du muscle de Müller. L’origine peut être myogène (fibrose, infiltration du muscle) ou encore neurogène (syncinésies de Marcus-Gunn sans ptosis associé par exemple). La rétraction congénitale de la paupière supérieure peut se rencontrer :
dans la maladie de Basedow congénitale par passage des anticorps maternels chez le fœtus (rare mais décrite);
le plus souvent, par fausse rétraction par un réflexe du type eye-popping-reflex. L’hypercontraction du muscle releveur de la paupière supérieure crée alors un aspect de rétraction lors de la baisse brutale de la luminosité. Celui-ci apparaît au cours du 3e mois et est spontanément résolutif vers l’âge de 8 mois. Il faut savoir l’expliquer et rassurer les parents;
dans les syndromes de Crouzon ou d’Apert ou bien s’intégrer dans un autre syndrome malformatif complexe. Ceux-ci s’accompagnent d’un micro-orbitisme, avec un aspect d’exorbitisme, de brièveté et la rétraction des quatre paupières [24]. En cas de luxation spontanée des globes oculaires avec menace sur leur intégrité lorsque les paupières passent en arrière du globe, une tarsorraphie précoce devra être réalisée. La chirurgie de la rétraction est rarement indiquée chez ces enfants. Les techniques d’allongement sont relativement comparables à celles réalisées pour l’adulte, à savoir une müllerectomie sans suture pour les formes faibles à modérées et des reculs du muscle releveur avec ou sans interposition pour les rétractions plus importantes;
la paralysie faciale congénitale : elle est la conséquence d’une agénésie congénitale du nerf facial. Elle peut être bilatérale et s’accompagner d’une atteinte de la motricité oculaire, comme dans le syndrome de Mœbius avec atteinte des nerfs crâniens VI et VII. Le visage est amimique avec une absence de tonus musculaire de l’orbiculaire palpébral rendant l’occlusion impossible. Seule la persistance d’un réflexe de Charles Bell permet une couverture satisfaisante du globe oculaire. Le but de la chirurgie est tout d’abord de protéger le globe oculaire et d’éviter des conséquences cornéennes. Pour cela le traitement est d’abord médical par l’application de pommade protectrice. Dans certains cas, un repositionnement de la paupière inférieure ou encore la mise en place d’une plaque d’or peut favoriser l’occlusion.
La tolérance cornéenne chez l’enfant est bien meilleure que chez l’adulte. L’intervention de repositionnement palpébral dans le cadre d’une paralysie faciale ne sera précoce que dans les risques oculaires majeurs.
Ces anomalies regroupent les malpositions du bord libre palpébral : épiblépharon, entropion, ectropion, malposition des canthi interne et externe.
L’épiblépharon correspond à un excès cutané de la partie médiale de la marge palpébrale entraînant une verticalisation de la ligne ciliaire. Il n’y a pas à proprement parler de rotation du bord libre, ce qui le distingue de l’entropion. Les cils sont orientés dans leur ensemble vers le haut avec un frottement non agressif, ce qui rend l’épiblépharon tolérable. Chez le Caucasien, il apparaît chez le nourrisson, du fait de la protubérance du massif jugal. Il peut entraîner un larmoiement avec des conjonctivites à répétition et constituer un diagnostic différentiel de l’imperméabilité du canal lacrymonasal. Chez les Asiatiques, l’épiblépharon est très fréquent, avec parfois même une atteinte de la paupière supérieure [25].
L’entropion congénital est beaucoup plus rare que l’épiblépharon, avec lequel il est cependant parfois confondu. L’entropion est le plus souvent associé à une pathologie de l’orbite ou une microphtalmie. Un œil de petite taille n’exercera plus une poussée dans le plan postéro-antérieur sur les paupières entraînant cet enroulement. L’entropion peut s’aggraver avec le temps. Les cils prennent une position horizontale, dirigée de manière agressive vers le globe oculaire, du fait de la rotation du bord libre, entraînant une kératite avec ses conséquences fonctionnelles à savoir un larmoiement, une photophobie. Dans certains cas, une ulcération cornéenne avec opacification tardive peut être à l’origine d’une amblyopie [26]. La distinction avec l’épiblépharon, qui a tendance à régresser spontanément, est importante à réaliser.
Comme chez l’adulte, l’entropion primitif est dÛ à une faiblesse du muscle rétracteur de la paupière inférieure. Les fibres de l’orbiculaire préseptales migrent alors en position prétarsale. On ne retrouve pas, contrairement à l’adulte, de laxité palpébrale.
Certaines affections congénitales ou même acquises de l’enfant peuvent entraîner un entropion de la paupière inférieure. Il est dÛ à une absence de soutien ou de pression de la lamelle postérieure palpébrale par un œil microphtalme par exemple, ou encore une prothèse mal adaptée. La mise en place de conformateurs de taille croissante comblant la cavité orbitaire permet de repositionner la paupière. Ils vont induire un développement palpébral et soutenir le bord libre. Dans certains cas, notamment chez les enfants ayant bénéficié d’une énucléation pour une pathologie maligne, un allongement de paupière inférieure par greffe conquale ou muqueuse palatine peut être nécessaire.
Ce syndrome particulier, décrit en 1948, correspond à un enroulement du bord libre de la paupière supérieure dÛ à une encoche naturelle longitudinale du tarse. Ainsi, un pli palpébral se crée au niveau tarsal, entraînant un enroulement de la paupière supérieure sur elle-même. Il s’agit d’une anomalie congénitale de formation du tarse lors de l’embryogenèse [27].
L’épiblépharon se corrige spontanément lors des premières années de vie chez le Caucasien. La croissance du massif facial et l’allongement du visage permettent un repositionnement favorable du bord libre avec la disparition du repli cutané pathognomonique de l’épiblépharon. Le traitement repose sur des soins locaux par lubrification de la surface. En cas d’ulcère, une intervention consiste à réaliser une excision modérée de l’excédent myocutané de la paupière inférieure associée à un repositionnement du sol ciliaire (eFig. 7-6).
Pour l’entropion, le traitement est plus complexe et repose sur une remise en tension des rétracteurs de la paupière inférieure. Pour l’entropion secondaire, l’allongement palpébral est parfois nécessaire. Le tarsal kink syndrom bénéficiera lui d’une suture éver-sante avec excision de la cicatrice tarsale au niveau de la paupière supérieure.
eFig. 7-6, 7f Technique par résection cutanée semi-lunaire
a. Dessin en regard de la zone d’entropion, b. Incision et résection semi-lunaire, c. Incision et résection semi-lunaire, d. Incision et résection semi-lunaire, e. Résection cutanée semi-lunaire pour la correction de l’épiblépharon, f.1 Suture éversante prenant peau – orbiculaire – tarse – orbiculaire et peau, f.2 Suture éversante prenant peau – orbiculaire – tarse – orbiculaire et peau
Comme chez l’adulte, l’ectropion congénital correspond à une éversion du bord libre d’une ou de plusieurs paupières, présent dès la naissance. Il peut être isolé primitif ou s’intégrer dans un syndrome de malformation plus complexe tel que le syndrome de blépharophimosis.
L’ectropion congénital isolé est exceptionnel et affecte le plus souvent les quatre paupières. On constate alors une brièveté de la lamelle antérieure palpébrale, plus ou moins importante, associée à une laxité du bord libre permettant la formation de l’ectropion. En cas de brièveté importante de la lamelle antérieure, des allongements par greffe cutanée sont parfois nécessaires. Elles sont de réalisation difficile chez un nourrisson qui présente peu d’excédents cutanés à prélever.
L’ectropion congénital associé est la conséquence d’une pathologie cutanée ou orbitaire.
L’ichtyose lamellaire ou bébé collodion est une forme grave. Le pronostic vital de l’enfant peut être engagé dans les formes graves. L’enfant présente à la naissance un aspect de grand brÛlé avec des troubles infectieux et métaboliques comparables. La rétraction cutanée intéresse l’ensemble du plan dermique, y compris les paupières entraînant un ectropion tractionnel.
Le blépharophimosis constitue un syndrome comprenant ptosis, épicanthus inversus, télécanthus et ectropion congénital.
La trisomie 21 comprend un ectropion des paupières inférieures, un épicanthus palpébral avec une inclinaison mongoloïde des fentes palpébrales. Elle est associée à des anomalies variées nombreuses du globe oculaire.
Les syndromes de fentes palpébrales décrites par Tessier peuvent entraîner un ectropion. La fente 6 correspond à une atteinte de la partie latérale de la paupière inférieure, jusqu’à l’éminence malaire. Le déficit est le plus souvent osseux, parfois associé à une hypoplasie des tissus mous et dans certains cas un colobome.
Dans le syndrome de Franceschetti, l’atteinte comprend une fente 6 bilatérale associée aux fentes 7 et 8.
Le traitement médical des ectropions congénitaux associés vise à sauvegarder la cornée est essentiel et repose sur une surveillance régulière et l’application d’agents mouillants et protecteurs telle la pommade vitamine A. Les traitements conservateurs, avec notamment lubrification de la peau et application d’émollients, seront indiqués pour les ichtyoses graves mais aussi pour certains cas de brÛlure ou des dermatites graves (eczéma).
Le traitement chirurgical est variable en fonction de l’anomalie retrouvée (eFig. 7-7). Le but est de restituer une certaine quantité de tissu palpébral de manière à éviter la traction du bord libre (eFig. 7-7). Il est indiqué en cas d’insuffisance de la lamelle antérieure avec exposition cornéenne (eFig. 7-8 et 7-9). En cas d’atteinte latérale, notamment pour le syndrome de Franceschetti, un allongement de paupière par lambeau de paupière supérieure à paupière inférieure à charnière externe peut être indiqué pour traiter ce cas d’euryblépharon (voir Fig. 7-3).
Néanmoins chez l’enfant, l’excès de paupière supérieure n’est pas toujours suffisant et on peut avoir recours à d’autres sites de prélèvement, tels que la région rétro-auriculaire ou encore sus-claviculaire (eFig. 7-10 et 7-11).
Il faut savoir que le résultat cosmétique à court terme sera moins bon, car la qualité de peau est différente et cela peut être visible chez l’enfant.
Dans certaines formes mineures avec rétraction de la lamelle antérieure peu importante, une canthopexie latérale de repositionnement du bord libre peut être indiquée.
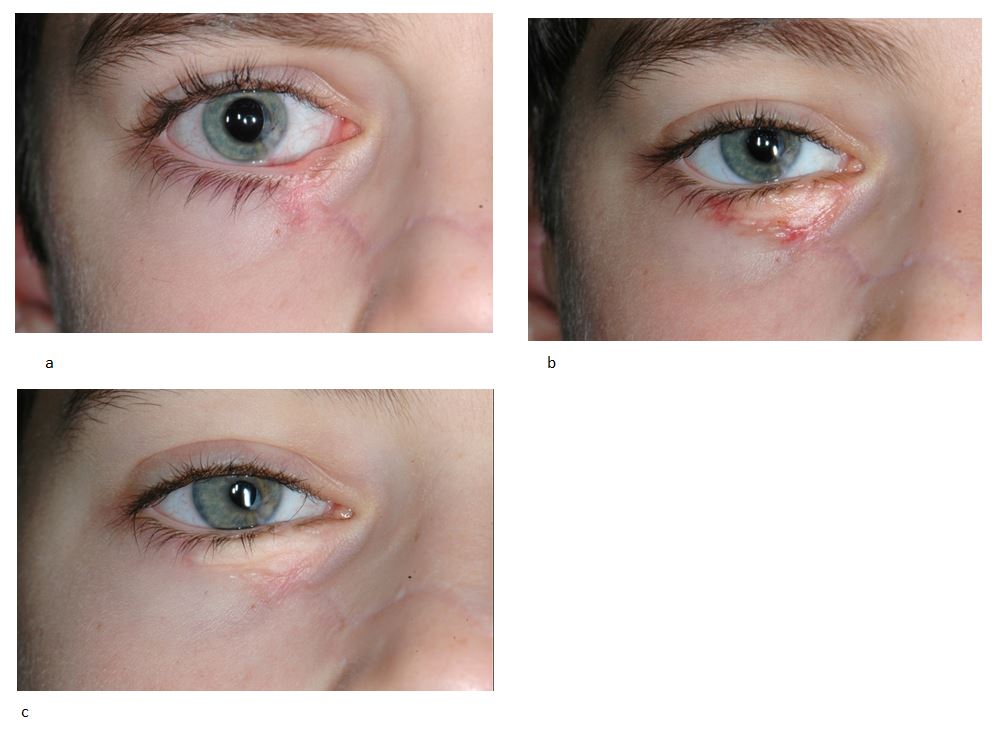
eFig. 7-7 Rétraction du bord libre palpébral post-traumatique, traitée par greffe de peau sus-claviculaire chez un enfant de 12 ans
eFig. 7-8 Suture bord à bord avec canthotomie, cantholyse de décharge
eFig. 7-9 Lambeau d’avancement par canthotomie, cantholyse
a. Ouverture du canthus externe, b. Section du chef inférieur du ligament canthal latéral, c. Décollement sous-périosté le long du rebord orbitaire inférieur facilitant la mobilisation, d. Suture du bord libre
eFig. 7-10 Coupe transversale de paupière inférieure distinguant lamelle antérieure et lamelle postérieure
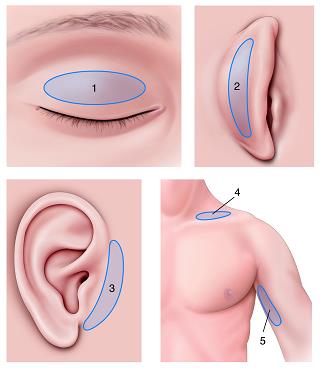
eFig. 7-11 Différents sites de prélèvement de greffe cutanée (a–d)
1. Dermatochalasis, 2. Rétro-auriculaire, 3. Prétragien, 4. Sus-claviculaire, 5. Face interne du bras
C’est une atteinte rare décrite par Desmarres pour la première fois. Elle consiste en une fente palpébrale plus large que la normale. Cette anomalie est le plus souvent symétrique avec un déplacement latéral et antérieur de la commissure externe.
L’association avec un ectropion de la paupière inférieure est fréquente du fait de cet excès de « bord libre palpébral ». Elle entraînera un épiphora avec risque d’exposition cornéenne.
La plupart des formes sont mineures et nécessitent une simple surveillance avec traitement lubrifiant. Pour les formes sévères avec ectropion et kératopathie d’exposition, le traitement chirurgical est alors indiqué. Il repose le plus souvent sur un raccourcissement du bord libre de la paupière avec canthopexie latérale permettant de réappliquer le bord libre au contact du globe oculaire [28]. En cas de brièveté de lamelle antérieure palpébrale associée, une greffe peut être indiquée.
Il s’agit d’une forme particulière de l’ectropion avec éversion bilatérale des paupières supérieures. Cette affection est le plus souvent transitoire, elle est due à un chémosis majeur de la conjonctive bulbaire et du fornix supérieur, lié à une obstruction du retour veineux au cours de l’accouchement lors d’une compression au passage de la filière utérovaginale. Elle atteint le plus souvent les nouveau-nés mélanodermes, nés de femmes primipares. On peut également la rencontrer lors de la trisomie 21.
Le traitement médical est local par lubrification cornéenne. Une compression exercée sur le globe oculaire maintenue par un adhésif peut permettre la régression du chémosis et de ses conséquences cornéennes [29].
Le traitement chirurgical est rarement indiqué, il consiste à réséquer, par voie postérieure, un lambeau conjonctival.
L’ensemble des éléments constitutif des paupières peut être le siège de développement d’une tumeur bénigne ou maligne. Seront évoqués les aspects les plus courants, ainsi que les lésions les plus graves.
L’épiderme malpighien palpébral constitue le principal revêtement de la paupière et ses lésions sont les plus fréquentes.
II s’agit d’une lésion exophytique, végétante pouvant se développer également sur le bord libre ou la conjonctive. Il s’agit d’une hyperacanthose (épaississement de la couche des cellules épineuses), une hyperpapillomatose (allongement des crêtes épidermiques et membrane basale sinueuse), ainsi que d’une hyperkératose (épaississement de la couche de kératine).
Fréquent chez l’enfant, il s’agit d’une lésion ombiliquée unique ou parfois multiple, avec un centre atteignant le versant cutané, le bord libre étant associé à une conjonctivite folliculaire (eFig. 7-12). Histologiquement, on retrouve une hyperplasie épidermique avec inclusions intracellulaires liées aux poxvirus. L’auto-inoculation par le grattage des lésions explique sa diffusion et le traitement consiste en l’exérèse chirurgicale des lésions.
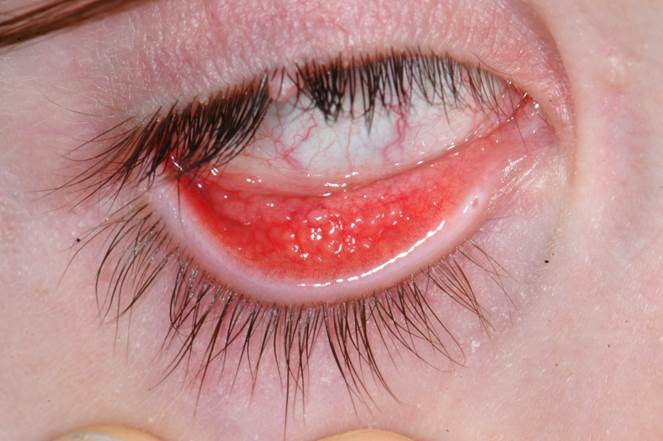
eFig. 7-12 Molluscum contagiosum du bord libre supérieur et conjonctivite folliculaire
Les premiers signes sont une sécheresse cutanée associée à un aspect tacheté de la peau dans les zones exposées au soleil, notamment au niveau de la face. On distingue trois stades de l’évolution :
premier stade : apparition au cours des deux premières années de la vie d’un érythème cutané avec desquamation et apparition des premières taches;
deuxième stade : des zones d’hypo- et hyperpigmentation et télangiectasies diffuses apparaissent, ainsi que des zones d’atrophie cutanée;
troisième stade : apparition de tumeurs malignes, les patients ont en effet un risque 2000 fois plus important que la normale de présenter un carcinome cutané. Les carcinomes basocellulaires ou épidermoïdes sont les plus fréquents. On peut cependant observer des mélanomes ou encore des carcinomes sébacés.
La maladie se transmet selon un mode récessif autosomique. Les signes cutanés sont précoces. L’évolution peut être, dans les cas les plus graves, rapide, avec lésions malignes cutanées dès l’âge de 3 ans. Le plus souvent, les lésions cutanées malignes apparaissent entre 8 et 10 ans. Dans la forme chronique et plus tardive, l’espérance de vie dépasse 20 ans ou plus. La prise en charge est d’abord préventive, reposant sur la protection solaire et l’exérèse des lésions suspectes de carcinome.
Chez l’enfant, les glandes sudoripares apocrines qui sécrètent un composant mucolipidique au niveau du bord libre palpébral sont le plus souvent responsables d’une lésion des annexes. Les hydrocystomes eccrines de petite taille prédominent au niveau des canthi. L’hydrocystome apocrine ou kyste de Moll est le plus souvent isolé, parfois pigmenté. Les syringomes peuvent s’observer dans la petite enfance. Ils sont également de petite taille, parfois non translucides, plus profonds, parfois confluents.
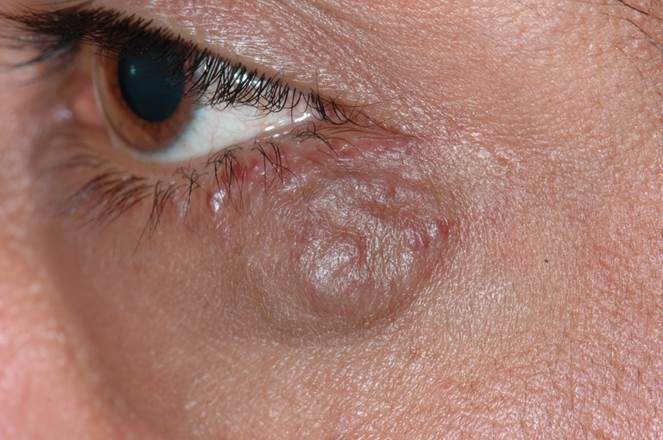
Le chalazion est l’atteinte la plus fréquente d’une glande sébacée. La glande de Meibomius au niveau du tarse palpébral présente une inflammation avec dilatation (eFig. 7-13).
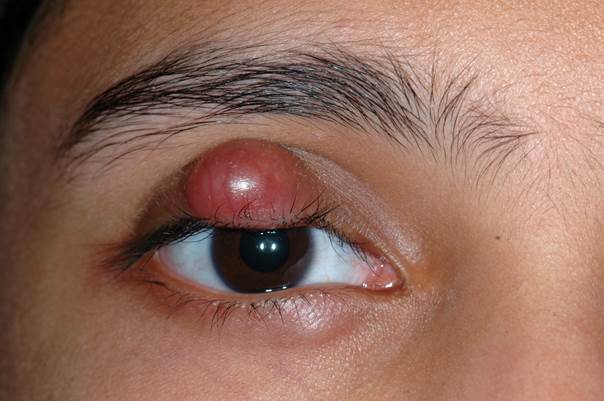
eFig. 7-13 Chalazion kystique de la paupière supérieure
Le traitement est tout d’abord médical reposant sur l’application de compresses ou d’un gant de toilette humide permettant une dilatation de l’ouverture de la glande, associée à des massages et l’application de pommade associant antibiotique et cortisone. En cas de résistance au traitement bien conduit, une exérèse chirurgicale peut être nécessaire. L’orgelet correspond à une infection bactérienne d’une glande meibomienne. Le kyste de Zeiss est une tuméfaction opaque d’une glande de Zeiss au niveau du bord libre (eFig. 7-14).
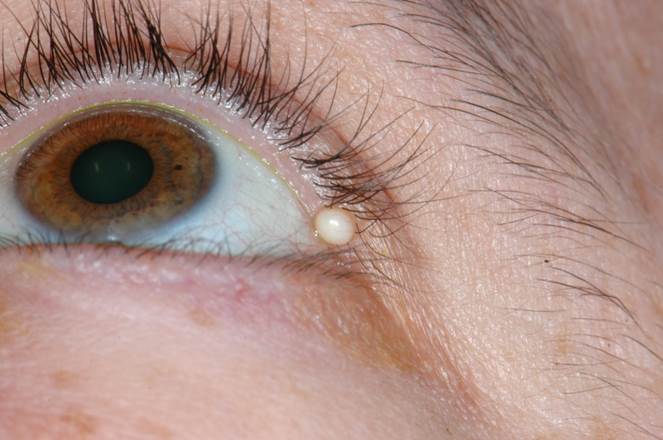
eFig. 7-14 Kyste de Zeiss
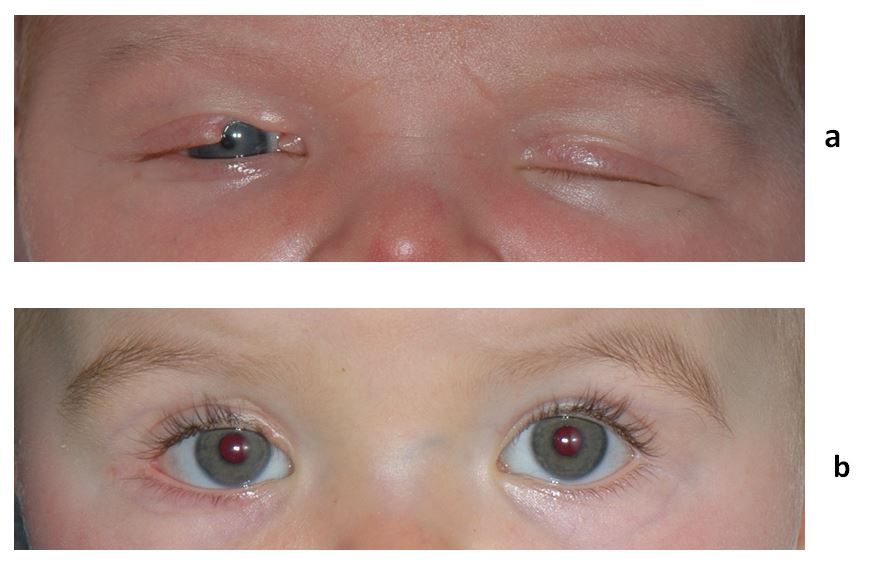
Les hémangiomes capillaires sont des hamartomes. Il s’agit de la malformation vasculaire palpébrale la plus fréquente chez l’enfant, le plus souvent sporadique, pouvant cependant parfois s’intégrer dans un syndrome complexe héréditaire. L’hémangiome est absent à la naissance et la lésion apparaît lors des premières semaines ou mois de vie. Elle grossit alors rapidement avec une couleur pourpre. L’examen retrouve un aspect cutané fripé, en regard de la lésion. Elle peut être superficielle, facilement reconnaissable, de couleur pourpre foncée avec une infiltration cutanée et une surface irrégulière. Dans certains cas, elle peut être profonde avec un respect du plan cutané et son diagnostic peut être plus difficile. En histologie, la tumeur est constituée de capillaires formés d’une couche unique de cellules endothéliales, les lobules étant contenus par un septum fibreux. L’évolution spontanée est une phase de croissance dans les 2 à 3 premières années de vie, puis une stabilisation et une régression à la fin de la première décennie. Dans les formes amblyogènes, le traitement s’impose, celui-ci repose dorénavant en première intention sur la prescription de bêtabloquants par voie systémique sous contrôle pédiatrique strict (Fig. 7-6). Dans certains cas, lors de lésions entraînant un ptosis mécanique, le recours à la chirurgie d’exérèse est parfois nécessaire (eFig. 7-15).
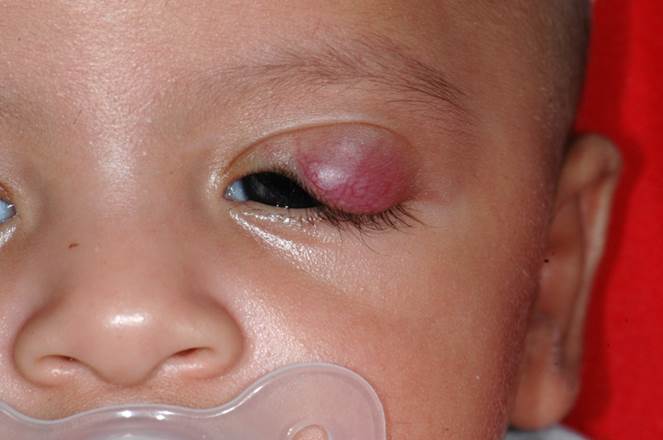
eFig. 7-15 Hémangiome capillaire infantile entraînant un ptosis et amputant l’axe visuel
Au contraire des angiomes capillaires, les angiomes plans peuvent être présents dès la naissance et résistent aux bêtabloquants. Ils feront suspecter la présence d’un syndrome de Sturge-Weber-Krabbe et justifieront la réalisation d’une imagerie cérébrale. Ils correspondent alors à un hémangiome de la face siégeant dans le territoire des deux premières branches du nerf trijumeau. L’examen retrouvera une hypertrophie de l’hémiface avec en imagerie cérébrale des calcifications intracrâniennes caractéristiques responsables d’une épilepsie.
Il s’agit d’un ensemble regroupant des atteintes hétérogènes. Pour les formes veineuses, la manœuvre de Valsalva est positive et l’examen retrouvera la présence d’un thrill vasculaire pour les lésions à haut débit vasculaire. Le traitement repose sur l’exérèse chirurgicale, précédée, dans certains cas, d’une embolisation (eFig. 7-16).
eFig. 7-16 Malformation artérioveineuse palpébrale caractérisée par un réseau veineux dilaté et une pigmentation bleuâtre
Il s’agit d’une tumeur nerveuse bénigne nodulaire, souvent multiple, dont le traitement est l’exérèse chirurgicale. En histologie, il s’agit d’une lésion composée de cellules de Schwann et de fibroblastes. On distingue le neurofibrome solitaire isolé du neurofibrome diffus beaucoup plus rare.
Le névrome plexiforme est la lésion pathognomonique de la neurofibromatose de type 1 ou maladie de Von Recklinghausen. Il apparaît dans l’enfance, grossit progressivement pour envahir la conjonctive et l’orbite. Sa consistance caractéristique en « pelote de ficelle » ou en « paquet de vers » est reconnaissable lors de l’intervention chirurgicale d’exérèse. Il entraîne une ptose de la partie latérale de la paupière créant un aspect en « S » caractéristique avec hypertrophie des différentes structures (eFig. 7-17). En histologique, le névrome plexiforme est composé d’une prolifération créant une gaine périnerveuse autour des nerfs périphériques.
eFig. 7-17 Névrome plexiforme de la paupière supérieure comblant le creux sustarsal et ptosant
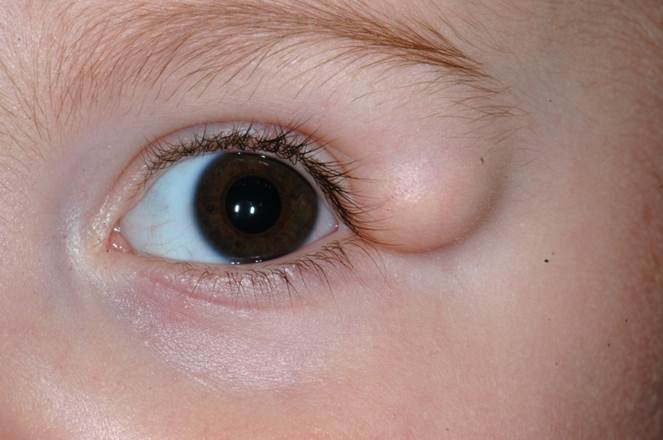
Les kystes dermoïdes sont limités par un épiderme constituant des annexes pilaires et sébacées. Le contenu est hétérogène. Ce sont des lésions limitées. Elles se situent le plus souvent au niveau de la queue ou de la tête du sourcil. Les kystes dermoïdes correspondent à des hamartomes, les éléments dermiques sont piégés lors de la suture des bourgeons du massif facial. Ainsi la localisation préférentielle du kyste dermoïde, dont l’origine est toujours profonde, est située à la suture de l’os frontal et de l’os zygomatique.
Le diagnostic est clinique. Une imagerie orbitaire est parfois nécessaire pour évaluer la position de la lésion. Le traitement est chirurgical, par un abord situé dans le pli palpébral (eFig. 7-18).
eFig. 7-18 Kyste dermoïde de la queue du sourcil
Le carcinome basocellulaire est la tumeur palpébrale maligne la plus fréquente des paupières. Elle se situe le plus souvent au niveau de la paupière inférieure, puis de la paupière supérieure et enfin au niveau des canthi. Histologiquement, les cellules basales prolifèrent avec des lobules et un aspect macroscopique perlé. Le syndrome de Gorlin se caractérise par l’apparition de basocellulaires au cours de l’adolescence ou chez le grand enfant. Le diagnostic repose sur l’âge d’apparition précoce, les antécédents familiaux et la présence d’au moins deux tumeurs. Le xeroderma pigmentosum peut présenter précocement, au cours de son évolution, des carcinomes basocellulaires et parfois même des lésions plus agressives de type carcinome épidermoïde.
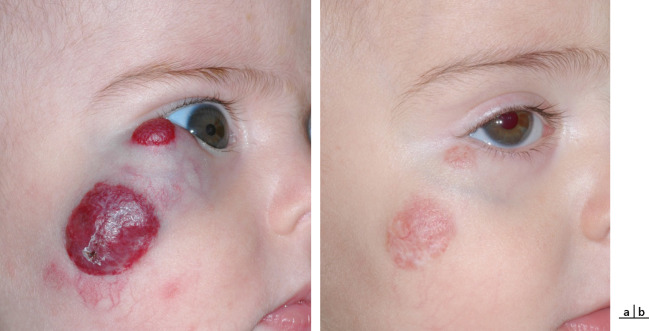
Fig. 7-6 Hémangiomes capillaires infantiles traités par bêtabloquant systémique. a b
Les atteintes palpébrales chez l’enfant sont une source d’inquiétude pour les parents.
De nombreuses questions se posent en effet :
L’œil est-il normal
L’enfant pourra-t-il voir?
Quelle est la cause?
Pourra-t-on corriger cette disgrâce et quand?
Le désarroi des parents est important du fait de cette anomalie faciale à laquelle ils n’étaient pas préparés. L’association à un syndrome malformatif rend la prise en charge difficile. Le chirurgien devra non seulement expliquer la physiopathogénie, son origine, mais aussi exposer les possibilités thérapeutiques et le pronostic fonctionnel et esthétique. Lorsque l’atteinte est modérée, la correction chirurgicale permet un résultat cosmétique satisfaisant. En revanche lorsque l’atteinte est sévère d’autant plus qu’elle est unilatérale, la symétrisation parfaite du regard dans toutes ses positions est impossible [7]. L’opérateur veillera bien à les prévenir de ce que l’on peut attendre de l’intervention et sur le fait qu’une nouvelle opération peut être nécessaire à l’issue de la croissance sans que cela ne soit le fait d’une complication ou d’une intervention mal réalisée. Un ajustement chirurgical en fin de croissance à la pré-adolescence, notamment en cas de ptosis sévère, est nécessaire. Chez le grand enfant et l’adolescent, les considérations cosmétiques viennent souvent au premier plan. La croissance faciale terminée, l’intervention réalisée sous anesthésie locale permettra un dosage peropératoire et ainsi un résultat cosmétique plus prévisible.
La chirurgie du ptosis, quelle que soit la technique, va entraîner une diminution du déroulé de la paupière vers le bas et une lagophtalmie (lid-lag). Le lid-lag diminue avec le temps sans disparaître totalement. Le réflexe de Charles Bell, s’il est satisfaisant, permettra lors du sommeil une protection de l’œil venant se placer en position supérieure sous la paupière. Si ce réflexe est de mauvaise qualité, notamment en cas de paralysie de l’élévation associée, l’opérateur veillera à minorer son geste chirurgical pour traiter le ptosis.
Les parents devront en être informés. L’instillation de pommade vitamine A, de manière répétée tout au long de la journée et au cours de la nuit, permettra d’éviter les atteintes cornéennes. Il est essentiel d’en informer l’entourage et de réaliser un apprentissage des soins à effectuer. Celles-ci peuvent parfois être graves avec des opacités séquellaires entraînant une amblyopie [30].
L’intervention chirurgicale est réalisée sous anesthésie générale, ce qui ne permet pas de réaliser un réglage peropératoire. Si la surcorrection est importante avec un risque cornéen, une réintervention précoce peut parfois être nécessaire. La sous-correction, beaucoup plus fréquente que la surcorrection, doit être un problème largement abordé avant l’intervention chirurgicale avec les parents. Elle doit être présentée comme, non pas une complication postopératoire, mais une étape dans la prise en charge du ptosis. Le praticien insistera sur le fait que le dosage peropératoire n’est pas possible, que la fonction du muscle releveur est parfois faible et que la réalisation d’un geste chirurgical, de résection maximale ou encore de suspension, ne permet malheureusement pas de restituer cette fonction. Il ne faut pas laisser les parents dans l’espoir d’une symétrisation parfaite qui ne sera pas possible.
Reformer le pli palpébral est indispensable pour les ptosis congénitaux. Une asymétrie supérieure à 1,5 à 2 mm de la distance entre le lit palpébral et le pli pourra justifier un éventuel repositionnement.
Une anomalie du contour palpébral est due à un mauvais positionnement des sutures unissant le muscle releveur au tarse pour les résections musculaires ou à un mauvais positionnement de la bandelette pour les suspensions frontales. Le contrôle du contour doit être nécessaire à chaque étape de la chirurgie. L’opérateur veillera à positionner correctement ses points, pour éviter tout aspect de « chapeau de gendarme », notamment lors des suspensions frontales.
Les difficultés de cicatrisation des paupières supérieures sont extrêmement rares chez l’enfant. Néanmoins, des granulomes ou des aspects inflammatoires peuvent se produire, notamment lors de l’utilisation de fils résorbables à dispersion lente pour les sutures cutanées.
Il sera important d’utiliser des fils à résorption rapide.
[1] Johnson CC Epicanthus and epiblepharon Arch Ophthalmol 1978 ; 96 : 1030-1033
[2] Mouriaux F, Hamedani M, Hurbli T, et al. Le syndrome de Waardenburg J Fr Ophtalmol 1999 ; 22 : 799-809
[3] Anderson RL, Nowinski TS The five-flap technique for blepharophimosis Arch Ophthalmol 1989 ; 107 : 248
[4] Mustardé JC The treatment of ptosis and epicanthal folds Br J Plast Surg 1959 ; 12 : 252
[5] Tazartes M Syndrome du centurion Les voies lacrymales. Rapport de la Société française d’ophtalmologie Paris: Masson (2006). 233-234
[6] Morris RJ, Collin JRO Functionnal surgery in Down’s syndrome Br J Ophthalmol 1986 ; 73 : 494-497
[7] Tessier P, Rougier R, Hervouet F Nouvelle classification anatomique des fentes faciales, craniofaciales et latérofaciales : leur répartition autour de l’orbite Chirurgie plastique orbito-palpébrale. Rapport de la Société française d’ophtalmologie Paris: Masson (1977). 192-208
[8] Clergeau G La réfraction de l’enfant Cahier de Sensori-Motricité 2008 ; 9 :
[9] Bok C, Berthout A, Galatoire O, et al. Le risque d’amblyopie dans le ptosis congénital isolé Pratiques en Ophtalmologie 2010 ; 38 : 2-4
[10] Anderson RL, Baumgartner SA Amblyopia in ptosis Arch Ophthalmol 1980 ; 98 : 1068-1069
[11] Hornblass A, Kass LG, Ziffer AJ Amblyopia in congenital ptosis Ophthalm Surg 1995 ; 26 : 334-337
[12] Dray JP, Leibovitch I Congenital ptosis and amblyopia : a retrospective study of 130 cases J Pediatr Ophthalmol Strabismus 2002 ; 39 : 222-225
[13] Harrad RA, Graham CM, Collin JRO Amblyopia and strabismus in congenital ptosis Eye 1988 ; 2 : 625-627
[14] Attebo K, Mitchell P, Cumming R, et al. Prevalence and cause of amblyopia in adult population Ophthalmology 1998 ; 105 : 154-159
[15] Anderson RL, Baumgartner SA Strabismus in ptosis Arch Ophthalmol 1980 ; 98 : 1062-1067
[16] Fiergang DL Wright KW, Foster JA Unilateral or asymmetric congenital ptosis head posturing and amblyopia J Pediatr Ophthalmol Strabism 1999 ; 36 : 74-77
[17] McCulloch DL, Wright K Unilateral congenital ptosis: compensatory head posturing and amblyopia Ophthal Plast Reconstr Surg 1993 ; 9 : 196-200
[18] Beneish R, Williams F, Polomeno RC, et al. Unilateral congenital ptosis and amblyopia Can J Ophthalmol 1983 ; 18 (3) : 127-130
[19] Foster JA, Katowitz JA Developmental eyelid abnormalities Pediatric oculoplastic surgery New York: Springer-Verlag (2002). 177-216
[20] Beard C Ptosis surgery : past, present and future Ophthal Plast Reconstr Surg 1985 ; 1 : 69-72
[21] Morax S, Benia L La suspension de la paupière au muscle frontal dans la chirurgie du ptosis. Technique et indications J Fr Ophtalmol 1986 ; 3 : 461-470
[22] Morax S Résection du releveur par voie cutanée dans la cure chirurgicale du ptosis J Fr Ophtalmol 1982 ; 5 : 249-255
[23] Wu SY, Ma L, Huang HH, Tsai YJ Analysis of visual outcomes and complications following levator resection for unilateral congenital blepharoptosis without strabismus Biomed J 2013 ; 36 : 179-187
[24] Collin JRO, Allen L, Castronuovo S Congenital eyelid retraction Br J Ophthalmol 1990 ; 74 : 542-544
[25] Ruban JM, Baggio E Chirurgie des malpositions palpébrales congénitales de l’enfant J Fr Ophtalmol 2004 ; 27 : 304-326
[26] Adenis JP, Robert PY Entropion, trichiasis et distichiasis. Encycl Med Chir Paris: Elsevier (2001). 14Ophtalmologie, 21-100-B-20.
[27] Demirel S, Firat C, Firat PG Modified temporary eyelid margin suture for correction of congenital horizontal tarsal kink : a novel surgical technique Ophthal Plast Reconstr Surg 2012 ; 28 : 300-302
[28] D’Esposito M, Magli A, Del Prete A Genetic study and surgical correction of euryblepharon Ophthalmologica 1979 ; 178 : 396-403
[29] Kronish J, Lingua R Pressure patch treatment for congenital upper lid eversion Arch Ophthalmol 1991 ; 109 : 767-8
[30] Morax S Ptosis et complications Pathologie orbito-palpébrale. Rapport de la Société française d’ophtalmologie Paris: Masson (1998). 226-53
B. Fayet, N. Moineau, D. Bremond-Gignac, E. Racy
Le plus souvent bilatéraux, les larmoiements par hypersécrétion sont, fait fondamental, accompagnés d’une sensation chronique de gêne oculaire, d’inconfort, de prurit, etc. Cette notion n’est pas toujours verbalisée par les enfants et il faut rechercher un plissement palpébral, une photophobie, etc. L’interrogatoire séparera les larmoiements chroniques des larmoiements aigus.
C’est essentiellement le fait des pathologies de surface comme les conjonctivites atopiques et les pathologies meibomiennes, etc.
L’entropion congénital est rare. L’épiblépharon, souvent peu gênant, va s’améliorer avec la croissance.
Les larmoiements aigus sont dominés par d’autres symptômes qui orientent le diagnostic vers l’étiologie.
Ils imposent une consultation ophtalmologique en urgence.
La kératoconjonctivite virale occupe une place à part en raison des risques de séquelles. La présence de vésicules devant le ou les canalicules s’observe dans l’herpès, le zona et la varicelle. Les vésicules font défaut dans les adénovirus, qui sont de loin les virus les plus fréquents.
À la guérison d’une kératoconjonctivite avec adénopathie prétragienne, l’attention doit être attirée par la persistance ou la réapparition brutale du larmoiement.
L’évolution de l’éventuelle canaliculite peut se faire vers une sténose canaliculaire, très rapidement progressive, dont le point de départ se situe 1 à 2 mm après le méat lacrymal.
Le traitement par les antiviraux est logique mais non évalué. L’intubation bi-canaliculo-nasale, lorsqu’elle est réalisée en urgence, donne 99 % de bons résultats. La sonde doit rester en place une année, et plus encore après herpès car les récurrences sont possibles. Au stade de séquelle, cette intubation bi-canaliculonasale n’est possible que lorsque la destruction canaliculaire n’est pas trop étendue (environ 50 % des cas). L’ultime recours sera la lacorhinostomie à la fin de l’adolescence.
Classiquement, ces larmoiements sont isolés et indolores. Le caractère unilatéral est très évocateur. La consultation ophtalmologique n’est pas urgente.
L’inspection et/ou l’examen à l’aide du biomicroscope feront le diagnostic.
Imperforation méatique :
l’anneau fibreux avasculaire est bien visible au sommet du tubercule lacrymal mais la lumière est recouverte par une pellicule translucide avasculaire. Elle laisse deviner par transparence la lumière lacrymale sous-jacente;
un aspect particulier est la stricturotomie congénitale : sous le voile tissulaire, on devine l’absence de coalescence des berges du canalicule.
Agénésie méatique et anomalie canaliculaire : les agénésies canaliculaires vraies sont rares. Les bifidités sont de découverte fortuite car rarement symptomatiques.
Fistules du sac lacrymal : elles sont localisées préférentiellement 1 ou 2 mm sous le tendon canthal médial (Fig. 7-7). L’abouchement cutané a l’aspect d’un anneau méatique. Dans la forme isolée, une larme perle épisodiquement au niveau de la région médiocanthale et s’accentue au moment des rhumes : l’œdème de la muqueuse nasale réduit ou supprime transitoirement l’évacuation lacrymonasale normale. Les larmes sont détournées vers la fistule qui devient alors progressivement fonctionnelle. À la guérison de la rhinite, le drainage va retrouver son cheminement préférentiel vers la fosse nasale et le larmoiement se tarira progressivement. Cela explique que les enfants soient peu gênés. La chirurgie se discute au cas par cas [1].
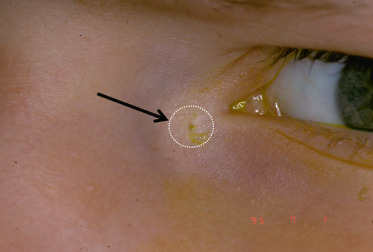
Fig. 7-7 Fistule congénitale du sac lacrymal.
Trois étiologies les résument principalement, la quatrième est plus anecdotique :
sténoses post-traumatiques : la réparation canaliculaire n’a pas suffisamment maintenu les extrémités sectionnées au contact l’une de l’autre (pas toujours simple, etc.). La cicatrisation de la plaie s’est faite avec une interposition d’un tissu fibreux plus ou moins important. Les réparations secondaires sont difficiles et d’autant moins couronnées de succès que la plaie est plus médiale;
sténoses post-virales : elles compliquent une kératoconjonctivite virale (voir plus haut);
sténoses iatrogènes [2] : elles se situent à la partie médiale du canalicule ou au niveau du canalicule commun. La réparation est délicate. Il faut souvent disséquer la zone sténosée avec réimplantation-suture dans le sac lacrymal. Le résultat fonctionnel est incertain. Dans ces trois situations, le sondage lacrymal localisera la ou les sténoses : il n’y aura pas de contact osseux si la sténose est complète. La distance sera mesurée à partir de l’anneau méatique qui sert de point de référence. L’imagerie n’est d’aucun secours le plus souvent;
paralysie faciale. Elles sont très rares chez l’enfant. Les problèmes posés sont comparables à celles de l’adulte. L’exploration instrumentale par sondage des voies lacrymales est normale.
Le préjudice des larmoiements isolés « clairs » chroniques est purement fonctionnel; il n’y a pas de risque d’infection.
Il n’existe pas de traitement médical.
L’importance du larmoiement est fonction de la perméabilité du canalicule homolatéral qui peut rester indemne.
Le rétablissement de la continuité lacrymale (conformateur méatique, intubation principalement bi-canaliculo-nasale, etc.) se fait chirurgicalement. Il faut qu’il soit anatomiquement possible, ce qui est peu prévisible avant l’exploration peropératoire.
La lacorhinostomie, ultime recours, est plutôt réservée à l’adulte.
Cette association signe quasiment le diagnostic d’imperforation lacrymonasale (ILN) congénitale lorsqu’elle est chronique. Les autres causes de sténose lacrymonasale (post-traumatique, fente faciale, etc.) sont très rares.
À l’interrogatoire, les parents signalent l’association d’un larmoiement chronique avec des sécrétions dont l’abondance est variable.
Elles sont de couleur crème en l’absence de surinfection.
Dans les formes mineures, elles se limitent à une simple sécrétion devant la caroncule.
Au maximum, elles agglutinent les cils. Le décollage à chaque réveil, malgré les instillations de sérum physiologique, tourmente autant les parents que l’enfant qui se laisse de moins en moins faire. Ces pseudo-conjonctivites réagissent bien aux antibiotiques, locaux ou généraux (donnés pour une autre cause).
Le plus évocateur n’est pas l’intensité des symptômes mais leurs récidives peu après l’arrêt de ces traitements. Après un certain temps d’évolution, la présence d’un eczéma de la paupière inférieure est classique (Fig. 7-8).
La distinction entre symptomatologie permanente et intermittente doit être recherchée avec insistance (voir plus loin). Les formes permanentes, mêmes variables en intensité, suggèrent un obstacle anatomique total. À l’inverse, les symptomatologies très intermittentes avec intervalles libres, francs, prolongés et indépendants de tout traitement, suggèrent un obstacle fonctionnel (obligatoirement muqueux). Ici, les symptômes sont volontiers déclenchés par un rhume, une éruption dentaire, etc. L’œdème de la muqueuse nasale va obturer transitoirement le canal lacrymonasal. Entre deux, le larmoiement est totalement absent. Si un test à la fluorescéine était réalisé, on verrait apparaître le colorant dans la fosse nasale. Pour les obstacles anatomiques, ces événements ne font qu’accentuer ce qui préexistait.
Lorsque les intervalles libres de toute symptomatologie sont de plus en plus fréquents et surtout de plus en plus durables et que progressivement les rhumes ne déclenchent plus le larmoiement, on peut imaginer que la croissance de la fosse nasale fait son œuvre.
Malheureusement, les tableaux cliniques ne sont pas aussi tranchés et l’association à un obstacle anatomique incomplet est possible.
De plus, les prescriptions d’antibiotiques locaux et/ou généraux, quelle qu’en soit la motivation, sont susceptibles de décapiter temporairement le tableau clinique, etc.
Dans la trisomie 21, il peut s’associer une rhinorrhée chronique qui traduit l’inflammation chronique de la muqueuse nasale.
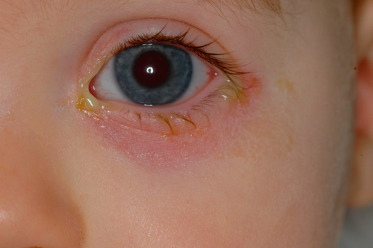
Fig. 7-8 Imperforation lacrymonasale avec eczéma de la paupière inférieure.
Une pression est exercée avec la pulpe d’un doigt sur la région médiocanthale, de bas en haut [3, 4]. Le déclenchement d’un reflux de mucus, même peu abondant, signe la sténose lacrymonasale. Son abondance est corrélée à l’importance de la dilatation du sac lacrymal (s’il n’avait pas été vidangé quelques minutes auparavant). Il n’est pas démontré à cette date une corrélation avec un type précis d’obstacle.
Une goutte de fluorescéine à 2 % est instillée dans le cul-de-sac conjonctival et l’on recherche des stigmates de son apparition en fosse nasale [5].
L’issue spontanée de colorant signe l’absence d’obstacle anatomique; on prendra bien garde que le colorant ne contamine pas l’orifice narinaire en s’écoulant de la fente palpébrale. La recherche de la présence du colorant sous le cornet inférieur à l’aide d’un coton-tige est très désagréable.
On s’enquerra de l’absence de pathologie générale, de reflux gasto-œsophagien, d’antécédents de trouble de l’hémostase, etc. Il est préférable que l’enfant ne soit pas enrhumé et à distance de son biberon. Il n’est pas démontré qu’un traitement local préalable modifie le pronostic.
Une anesthésie par collyre anesthésiant est discutable : son efficacité antalgique au niveau lacrymal n’est pas certaine.
On repère l’anneau méatique au sommet de son tubercule lacrymal grâce à sa couleur nacrée. Il est béant spontanément. On utilise une sonde lacrymale à bout mousse. Le diamètre est en moyenne de 0,3 mm en haut et 0,5 mm en bas. Il peut être nécessaire de le dilater préalablement, surtout en cas de blépharite ou pathologie de surface, car l’inflammation chronique tend à réduire son diamètre.
Trois étapes sont ensuite à respecter :
étape horizontale, canaliculaire : la sonde lacrymale est introduite par un canalicule et guidée jusqu’à rencontrer la gouttière lacrymale. Cette sensation est le « contact osseux » (1). Une traction latérale douce sur la paupière peut faciliter l’obtention du contact osseux. Cette étape est stressante et désagréable mais peu douloureuse à l’inverse des suivantes;
étape de rotation : durant cette phase, la paupière ne doit plus être en traction et le contact osseux ne doit jamais être relâché. Une rotation médiale à 90° est imprimée tout en rasant le sourcil. Au cours de ce geste, la paupière va venir se plaquer contre la région médiocanthale. Elle ne doit manifester aucun signe d’abaissement. Si tel était le cas, il faudrait immédiatement interrompre le sondage et recommencer à son tout début (signe de la fausse route). Le choix du canalicule supérieur ou inférieur est sans grande importance pratique puisque l’on veut explorer le sac lacrymal. Le canalicule inférieur est plus aisé à cathétériser mais l’étape de rotation est un peu plus simple lorsque l’on est passé par le canalicule supérieur. La présence d’une stricturotomie congénitale inférieure risque de se déchirer totalement lors de cette rotation : il faut soit bien accompagner la paupière soit préférer le canalicule supérieur;
étape verticale, lacrymonasale : en fin de rotation, la sonde va déraper le long de la gouttière. Habituellement, après un intervalle libre de 1 cm environ, la sonde rencontre un autre obstacle, de consistance un peu élastique (contact muqueux; Fig. 7-9, 2). Cette sensation tactile est associée à la description de l’imperforation de la valve lacrymonasale (dite de Hasner). Une accentuation de la pression sur la sonde permet de franchir aisément ce diaphragme muqueux (s’il était présent). La course de la sonde est de nouveau « libre » jusqu’à rencontrer le plancher de fosse nasale (contact nasal; Fig. 7-9, 3). L’axe de la sonde est le plus souvent en bas et un peu en dedans.
Pour certains, le sondage lacrymal doit être complété par la recherche du contact métallique (Fig. 7-9, 4) à l’aide d’une deuxième sonde nasale, large et à bout mousse, guidée sous le cornet inférieur à la recherche de la précédente [6]. On tentera de mobiliser la première sonde lacrymale en passant de part et d’autre. Le contact métallique positif suggère que le sondage a été bien conduit et que la sonde lacrymale est libre dans le méat nasal inférieur (Fig. 7-9). La sensation tactile doit être très franche. L’absence de contact métallique est anormale. Elle signe pratiquement le trajet sous-muqueux. Rappelons que les études endoscopiques [7] ont montré que dans 10 % des cas la sonde nasale peut mobiliser la sonde lacrymale sans que celle-ci ait totalement perforé la muqueuse qui a été simplement repoussée par la sonde haute. Le sondage peut être plus laborieux, soit parce que l’enfant est d’une vigueur inhabituelle soit parce que l’anatomie n’est pas assez compliante. Primum non nocere.
Il est préférable de renoncer temporairement et programmer un nouvel examen quelques semaines plus tard, au besoin sous anesthésie en fonction de l’âge de l’enfant (voir plus loin).
Après sondage lacrymal, l’énoncé aux parents d’un pronostic gagne dans tous les cas à être prudent.
Les complications graves du sondage lacrymal sont rarissimes. Un petit saignement, lacrymal et/ou nasal, peut s’observer. Il est sans aucune gravité. Les spasmes du sanglot sont aussi bénins qu’ils sont impressionnants.
Il ne faut pas redresser et réemployer les sondes lacrymales tordues en raison du risque de fragilisation et de rupture intralacrymale lors d’un examen ultérieur.
Il est habituel de prescrire des instillations de collyres associant dexaméthasone et antibiotiques pour une durée de 1 semaine [8 - 9]. L’adjonction de mitomycine lors du sondage n’a pas encore été évaluée [10]
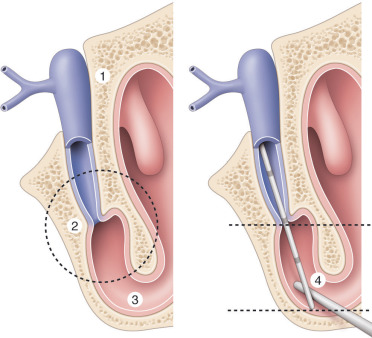
Fig. 7-9 Résumé du sondage lacrymal et des quatre sensations tactiles.
Pour certains, on pourrait toujours réaliser une contention mécanique qui s’effectuerait en ce cas en emmaillotant solidement (momification) le nourrisson, éventuellement en cabinet médical [8, 9, 11, 12]. Cela serait possible jusqu’à l’âge d’environ 12 mois, Au-delà, il va être malaisé d’immobiliser efficacement la tête, l’exploration instrumentale devenant plus laborieuse et potentiellement moins efficace, voire délétère. En pratique donc, le sondage du nourrisson sans anesthésie est à éviter et à abandonner.
L’anesthésie se déroule au bloc opératoire. La présence du masque facial ne gêne quasi pas l’accès aux voies lacrymales.
Il existe deux niveaux d’anesthésie générale :
l’anesthésie en ventilation spontanée à l’aide d’un masque facial est suffisante pour un examen ophtalmologique, un sondage lacrymal ou une intubation poussée [13];
l’anesthésie générale en ventilation mécanique assistée. Elle prolonge la première étape. Elle permet la mise en place d’une protection laryngée. Elle est indiquée pour des chirurgies longues et/ou à potentialité hémorragique peropératoire (intubations tirées, intubation rétrograde, dacryocystorhinostomie [DRC]).
Le risque de complication générale augmente avec l’importance de l’anesthésie générale.
Signalons la possibilité d’une anesthésie/sédation par inhalation de protoxyde d’azote en consultation [14] qui faciliterait l’exploration instrumentale (moins de douleur). Nous n’en avons pas l’expérience.
L’extrémité de la sonde rencontre un obstacle muqueux environ 1 cm après le contact osseux.
Lorsque les sensations tactiles sont différentes, il faut envisager une variante anatomique et/ou une fausse route. Quatre structures participent, à des degrés divers, à la constitution d’une sténose lacrymonasale : le maxillaire, le cornet inférieur et les muqueuses lacrymale et/ou nasale. Jones [5] décrit 9 variétés d’imperforations lacrymonasales qui résultent de la combinaison de ces différents éléments. Sa description fait autorité dans la littérature lacrymale. II n’est pas aisé de retrouver des travaux anatomiques qui corroborent ses conclusions. Cette région anatomique est très difficile d’accès et livre laborieusement ses secrets. Même en l’absence de corrélations anatomocliniques, on peut imaginer qu’il existe un continuum lésionnel entre au maximum les agénésies osseuses et les imperforations d’un simple vélum muqueux. Ces dernières sont de très loin les plus fréquentes.
La rhinoscopie antérieure se déroule au bloc opératoire avec une sédation et toutes les précautions anesthésiques d’usage et pour certains, en collaboration avec un ORL. Le décongestionnement de la muqueuse nasale (Xylocaïne® adrénalinée 1 % , etc.) est rarement suffisant pour observer l’abouchement du canal lacrymonasal. La subluxation du cornet inférieur est le plus souvent indispensable. La rhinoscopie ne renseigne que sur la valve lacrymonasale des voies lacrymales d’excrétion (VLE) et méconnaît tout ce qui se passe en amont. Ce contrôle visuel permet dans une certaine mesure de rectifier l’axe du cathétérisme, ce qui améliorerait les performances du sondage [7]. La rhinoscopie n’a pas sa place en routine.
Le lavage à la canule, pour mémoire, est peu employé chez l’enfant car on redoute de déclencher un spasme bronchopulmonaire en réaction à une inhalation de sérum physiologique si l’enfant n’est pas intubé avec une sonde à ballonnet. L’endoscopie intralacrymale reste au stade de recherche. La radiographie avec opacification est pratiquement abandonnée. L’imagerie actuelle (scanner et IRM) est peu contributive compte tenu de la petite taille des structures. Seul un dacryoscanner, c’est-à-dire avec injection de produit de contraste dans les voies lacrymales, peut permettre de les visualiser parfaitement; les indications sont rares.
La symptomatologie peut disparaître spontanément définitivement. Cette courbe de guérison naturelle décroît régulièrement, 60 à 90 % selon les études [5] environ se faisant au cours des 6 premiers mois comme le rapportait notamment le Pediatric Eye Disease Investigator Group en 2012 sur une série de 133 yeux. Au-delà du 12e mois, les guérisons spontanées des formes permanentes deviennent rares [15].
La symptomatologie peut persister sur un mode simple. Cela peut entraîner un retentissement social. Le personnel des collectivités redoutant une conjonctivite virale contagieuse répugne à accueillir l’enfant.
La symptomatologie peut s’accentuer sous forme d’une dilatation permanente du sac lacrymal. Cela est rare chez l’enfant. Les raisons qui font évoluer l’imperméabilité vers une persistance simple à « sac plat » ou vers une mucocèle lacrymale (sac lacrymal dilaté) ne sont pas parfaitement connues (Fig. 7-10).
Hormis le préjudice social, les complications sont plutôt exceptionnelles : la plus fréquente étant l’eczéma des paupières et la plus rare, l’abcès du sac lacrymal.
Au long cours, les instillations de collyres bactériostatiques atténuent les sécrétions.
Ils sont préférables aux collyres bactéricides car sans risque de sélection de germes.
Les frottis sont sans grand intérêt pratique, tant que l’œil reste blanc, etc.
Il n’existe pas de traitement médical des ILN. Il faut mécaniquement rétablir la continuité lacrymale.
Les principes thérapeutiques sont multiples.
Ce massage est rassurant et présente le mérite de pouvoir associer activement la famille à la thérapeutique. Quelques études semblent démontrer l’efficacité par rapport à l’abstention, ainsi Karti [4] rapporte, sur une cohorte de 31 enfants âgés de moins de 1 an ayant été traités par massages réguliers (n = 24), 91,6 % de succès à 1 an contre 77 % sans massage régulier (n = 8). Il souligne l’importance du rôle des parents dans cette prise en charge non chirurgicale [7, 8].
Ses partisans formulent l’hypothèse que la pression exercée sur la région médiocanthale se transmet à la valve lacrymonasale, au point de provoquer sa déchirure, etc.
Les observations rhinoscopiques ne confirment pas cette transmission lorsque le sac lacrymal n’est pas distendu.
Soulignons que la période où le taux de guérisons spontanées est le plus élevé correspond à celle où le massage antérograde du sac est mis en œuvre. C’est pourquoi, pour d’autres auteurs, massage du sac ou abstention sont équivalents.
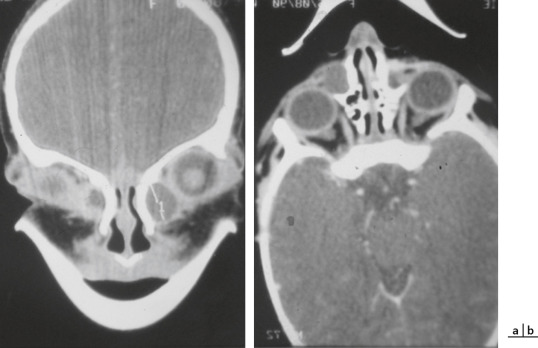
Fig. 7-10 a, b. Distension néonatale du sac lacrymal. Imagerie du sablier lacrymal.
(Remerciements au Pr. D. Denis.)
1. Par sondage. L’intérêt thérapeutique du sondage est de déchirer la muqueuse lacrymale et/ou nasale. L’expérience clinique démontre l’efficacité thérapeutique remarquable des sondages lacrymaux dans les formes simples [8, 9].
Certains facteurs pronostiques permettent d’en évaluer l’intérêt et les probabilités de réussite [17] :
âge au moment du sondage (plus on avance en âge, moins le taux de réussite est important);
antécédent/échec de sondage antérieur (moindre taux de réussite);
en peropératoire : sondage simple ou laborieux.
Les échecs du sondage peuvent s’expliquer par trois mécanismes :
sténoses complexes;
trajet sous-muqueux (15 % ). Le sondage lacrymal sous guidage endoscopique permettrait d’améliorer ce score [18];
la muqueuse cicatrice et la sténose se reproduit.
Que faire en cas d’échec d’un premier sondage? Il n’est pas illogique de faire une nouvelle tentative quelques semaines plus tard, même si la sensation du premier sondage ne laissait guère d’espoir, la croissance pouvant modifier l’anatomie parfois de façon surprenante. Si deux, voire trois tentatives, n’ont pas réglé le problème, il est douteux qu’une nouvelle tentative soit enfin couronnée de succès [19].
2. Par intubation. Elle jouerait un rôle de stent et/ou d’aérateur empêchant la re-sténose et en guidant l’épithélialisation. Les intubations poussées adoptent le principe du cathéter veineux, ce qui dispense de l’étape de récupération nasale (par exemple Masterka™). La même intendance anesthésique que celle d’un sondage tardif suffit [13]. Les intubations tirées imposent une protection laryngée. La récupération du guide de pose (mandrin, fil guide par technique de Ritleng®) en fosse nasale étant potentiellement hémorragique, elle peut se faire avec ou sans contrôle visuel [18, 20].
3. Par dilatation à l’aide d’un ballonnet gonflable [21, 22]. Il s’agit d’une adaptation des sondes vasculaires d’embolectomie aux voies lacrymales. L’influence de cette dilatation surajoutée par rapport à une déchirure muqueuse simple n’est pas encore démontrée. La sonde est coÛteuse et la plus-value pour le patient mérite d’être éclaircie.
Le cornet inférieur est saisi près de sa racine d’insertion sur la paroi latérale de la fosse nasale à l’aide d’une pince. Le mouvement de torsion est imprimé à la pince pour provoquer la subluxation. Le contrôle visuel est difficile en raison du peu de place pour le passage simultané de deux instruments et du saignement fréquemment induit. Cette subluxation augmenterait la largeur du méat nasal inférieur et/ou mobiliserait positivement la partie de cornet inférieur entrant dans la constitution du canal lacrymonasal. Aucune de ces deux hypothèses n’a jamais été démontrée, pas plus que le devenir de cette subluxation lorsque s’épuise l’effet des vasoconstricteurs quelques heures plus tard. La subluxation du cornet inférieur est le plus souvent associée dans le même temps à d’autres gestes, et il est bien difficile de savoir en cas de succès lequel a été efficace.
Cette intervention chirurgicale établit une anastomose entre la cavité lacrymale et le méat nasal moyen. Cela court-circuite le conduit lacrymonasal. La DCR comporte une ostéotomie de la gouttière lacrymale puis un temps muqueux qui marsupialise tout le sac lacrymal dans le méat nasal moyen adjacent. Elle peut être envisagée par voie canthale médiale ou endonasale, si la fosse nasale est assez large (en pratique vers l’âge de 5 ans). Ce by-pass (dérivation) est le recours dans les anomalies osseuses irréductibles et pour les échecs d’intubation.
Insufflation d’air sous pression, ostéopathie, résection muqueuse sous le cornet inférieur, etc. Nous n’en avons pas l’expérience.
De prime abord, il semble déconcertant que le traitement d’une situation aussi banale ne soit toujours pas consensuel et que les résultats rapportés pour une même technique puissent varier de 30 à 100 % [24, 25]. Les séries sont difficilement comparables en raison d’importants biais de sélection :
un larmoiement n’est qu’un symptôme. Il est bien difficile à quantifier. La sécrétion lacrymale varie d’une heure à l’autre et est totalement indépendante de l’élimination, etc.;
anatomiquement, les ILN sont plurielles et les associations de sténoses sont possibles, etc.;
il faut prendre en compte la courbe de guérison naturelle avec le temps [15, 26];
les associations techniques mélangent les effets des unes avec les autres (l’intubation classique comporte une subluxation a minima, la subluxation thérapeutique comporte un sondage, etc.);
il existe une insuffisance d’études cliniques prospectives randomisées conséquentes [27];
on déplore la rareté des corrélations anatomiques et rhinoscopiques;
enfin, les rapports sociétaux (offre de soins, générosité des systèmes de santé, pression médico-légale, etc.), très différents d’un pays à l’autre, influencent subrepticement les comportements. Cet aspect peu étudié est probablement plus important qu’il n’y paraît au premier abord.
Malgré l’absence de consensus, trois grandes attitudes se distinguent, chacune se fondant sur une hypothèse physiopathogénique propre.
Certains auteurs postulent que la croissance va arranger la très grande majorité des cas. Après 4 ou 5 années de patience ne subsisteront que les vraies sténoses. Ses promoteurs mettent en avant 95 % de bons résultats sans jamais traiter par excès (coÛt, iatrogénie, etc.). On lui reproche un taux de DCR très élevé. Cette école est assez consensuelle en Europe du Nord.
D’autres auteurs postulent, à l’inverse, que l’inflammation chronique traînante aggrave avec le temps les désordres anatomiques. Leur sélection thérapeutique se fait en fonction de l’âge de l’enfant au moment de la consultation : sondage thérapeutique à partir du 6e mois, intubation lacrymonasale à partir du 12e mois et DCR pour les rares échecs et/ou impossibilités d’intubation tirée. Ses promoteurs mettent en avant 98 % de bons résultats à chaque tranche d’âge. Cette stratégie présente le mérite de ne pas faire perdre de temps et un pourcentage de DCR dérisoire (< 1 % ). À cette date, cette école est la référence particulièrement en France et aux États-Unis [28].
Enfin, certains postulent que la sténose lacrymonasale est invariable : un désordre anatomique simple ne s’aggravera pas avec la croissance et inversement. Leur choix thérapeutique se fait après un sondage exploratoire sur table systématique. Il arbitrera sans idée préconçue :
sténose simple : traitement simple (sondage tardif) avec une intendance anesthésique simple (anesthésie générale en ventilation spontanée) jusqu’à l’âge de 5 ans [29 - 34];
sténose complexe (et échec des précédents traitements) : intubation lacrymonasale avec une anesthésie générale en ventilation mécanique assistée et protection laryngée.
Faute de consensus, les décisions se prennent au cas par cas, en tenant compte de l’environnement familial, des traitements antérieurs, des possibilités techniques et anesthésiques ainsi que des habitudes de chaque opérateur. Dans tous les cas, le pronostic est moins bon dans les trisomies 21 en raison des pathologies nasales associées [35, 36].
Notre attitude est résumée dans la Fig. 7-11.
Il s’agit d’une variété d’ILN de l’enfant [37]. Plusieurs noms désignent cette pathologie néonatale du sac lacrymal : amniocèle, amniotocèle, mucocèle congénitale, dacryocèle, dacryocystocèle, kyste lacrymal congénital ou, pour nous, dilatation néonatale du sac lacrymal.
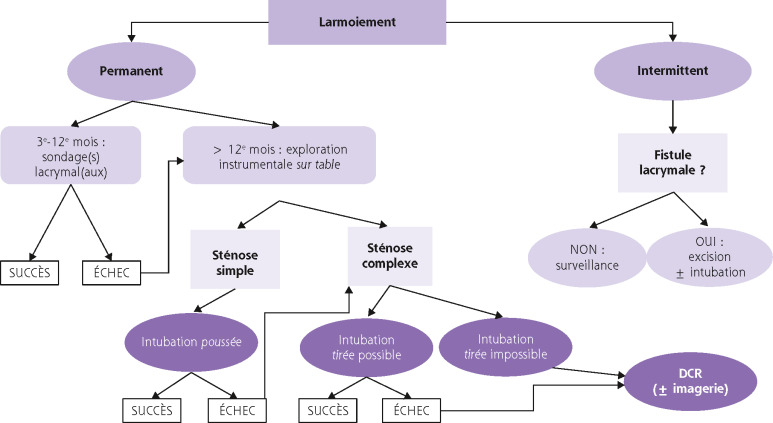
Fig. 7-11 Algorithme des indications et choix thérapeutiques des sténoses lacrymonasales.
N.B. : intubation poussée = type Masterka™ ; intubation tirée = par technique de Ritleng® par exemple.
Normalement, l’ébauche lacrymale embryonnaire se tunnelise d’un seul tenant, du méat à l’ostium, dès la 25e semaine d’aménorrhée. Les battements palpébraux sont présents et efficaces vers la 24e semaine. Le liquide amniotique pénètre à l’intérieur des voies lacrymales d’excrétion et peut probablement y circuler.
La coexistence d’une imperforation lacrymonasale et d’une hypercontinence de la valve canaliculosacculaire séquestre le liquide amniotique dans la cavité lacrymale qui se distend dans son ensemble. Les cas les plus précoces ont été rapportés dès les 30e et 32e semaines d’aménorrhée [37].
La cavité lacrymale peut se distendre extrêmement rapidement, en quelques jours ou quelques heures seulement. Cette distension lacrymale globale reste irréductible tant que perdure l’obstacle lacrymonasal.
Le canal lacrymonasal segmente cette distension lacrymale en deux parties distinctes mais qui restent fondamentalement en continuité : la partie canthale et le sac lacrymal lui-même. La partie nasale prend l’aspect d’une poche muqueuse qui comble le méat nasal inférieur. La poche nasale médialise le cornet inférieur peu ossifié à cet âge.
Il ne s’agit pas d’un kyste au sens histologique car cette dilatation est dépourvue de paroi propre. Sa paroi est constituée de muqueuse nasale normale.
Les caractéristiques des images échographiques (topographie, réflexivité, etc.) du sac lacrymal dilaté permettent d’éliminer les pathologies congénitales comme l’angiome, le lymphangiome, le méningocèle ethmoïdale ou un tératome. Le contenu de la cavité présente une échostructure (réflexion) très différente de celle du tératome et des angiomes.
Sa paroi n’est pas significativement vascularisée (Doppler à codage couleur).
Les dilatations lacrymales in utero sont plus fréquentes (environ 5 % ) que ce que l’on constate à la naissance (2 % ). En effet, il existe des guérisons spontanées in utero (et parfois des récidives).
L’accouchement par voie basse est susceptible de guérir un bon nombre de cas.
La distension médiocanthale est constatée dès la naissance, l’apex de la distension se situe sous le tendon canthal médial. Le creux sus-tarsal n’est pas comblé. Sa coloration est bleutée pseudo-angiomateuse (Fig. 7-12a). La palpation digitale perçoit un caractère liquidien rénitent. Aucun reflux de mucus ne se produit par les canalicules.
Un examen rhinoscopique antérieur visualise sous le cornet inférieur, plus ou moins médialisé, une protrusion qui comble le méat nasal inférieur (Fig. 7-12b). La pression sur l’une augmente la protrusion de l’autre et réciproquement, démontrant que ces deux cavités, canthale et nasale, communiquent l’une avec l’autre. Cela justifie à nos yeux l’appellation de « sablier lacrymal ».
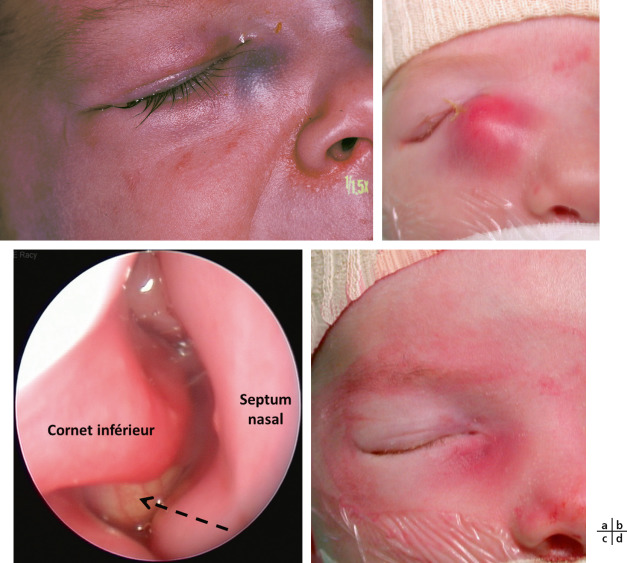
Fig. 7-12 Distension néonatale du sac lacrymal.
a. Forme simple. b. Forme abcédée. c. Vue endoscopique, expansion nasale. d. Après traitement.
(Fig. b et d : remerciements au Pr. D. Denis.)
Surinfection (dacryocystite). Dans 20 % des cas, se produit une surinfection du contenu lacrymal. La coloration bleutée de la tuméfaction est remplacée par une couleur rouge (Fig. 7-12c, d). La température est normale. L’évolution vers une fistulisation sacculocutanée est exceptionnelle. Bien que spectaculaire, le pronostic esthétique est excellent.
Détresse respiratoire néonatale : apanage des formes bilatérales et extensives. Jusqu’à l’âge de 2 ans l’enfant est un respirateur nasal exclusif et le flux d’air passe à 95 % par la partie basse de la fosse nasale. Cette double obstruction est comparable à celle d’une atrésie des choanes. Les formes bilatérales dépistées par l’échographie in utero permettent d’établir préventivement un plan de soins adapté.
Pour les formes simples, la guérison spontanée s’observe dans 80 % des cas et au cours du premier mois. La récidive après guérison spontanée est rare. Dans ces récidives, le liquide n’est plus de nature amniotique mais de simples larmes. De ce fait, le terme d’amniocèle pour désigner cette pathologie néonatale du sac lacrymal paraît un peu réducteur. Le massage énergique du sac trouvait chez les anciens toute sa légitimité. Le sondage lacrymal a ses adeptes. Il semble douteux qu’il soit intrinsèquement efficace sur une paroi nasale aussi médialisée, l’axe de la sonde ayant de chance d’être transfixiant.
Le traitement des formes compliquées repose sur la physiologie du sablier lacrymal (voir pour résumer Fig. 7-13). La fosse nasale est tamponnée par une mèche imbibée de Xylocaïne® adrénalinée. La poche nasale est repérée sous guidage endoscopique puis ponctionnée sous contrôle de la vue. Cela affaisse le sac lacrymal distendu. L’antibiothérapie générale n’est pas indispensable. Une vidéo de la technique peut être retrouvée sur Internet2.
Dans certaines distensions néonatales du sac lacrymal, les « poches nasales » sont parfois moins visibles à la partie antérieure de la fosse nasale en raison d’un cornet inférieur peu médialisé (cause ou conséquence?). Le développement de la poche nasale est alors plus postérieur. Une subluxation complémentaire du cornet inférieur peut être utile.
L’aspect nasal réel des différentes variétés d’ILN est encore mal connu. Là encore, il est vraisemblable qu’il existe un continuum lésionnel entre une paroi muqueuse totalement latéralisée, et plaquée contre la paroi latérale de la fosse nasale d’un côté, et une paroi muqueuse soufflée et totalement médialisée.
Dans la forme typique, scanner et IRM ne semblent pas indispensables au diagnostic.
La méningo-encéphalocèle se présente comme une tuméfaction qui part de la région frontale et comble le creux sus-tarsal; La poche nasale, lorsqu’elle descend aussi bas, plaque le cornet inférieur contre la paroi latérale et non contre la paroi médiale. Les pathologies nasales congénitales (hétérotopies, etc.) sont très différentes.
Les dacryocystites chroniques sont très rares chez l’enfant. L’expérience indique que les ILN négligées n’évoluent pas obligatoirement avec le temps vers une dilatation du sac. Cette dilatation est lente à se constituer à l’inverse des sténoses lacrymonasales traumatiques qui évoluent vers une dilatation sacculaire parfois même en quelques jours.
Une fois constituée, la dilatation du sac lacrymal est comparable en tout point à celle de l’adulte.
Le traitement n’est pas consensuel. C’est l’indication pour certain d’une DCR d’emblée. Pour notre part nous préférons prendre notre décision après avoir testé la résistance de l’obstacle lacrymonasal lors du sondage sur table.
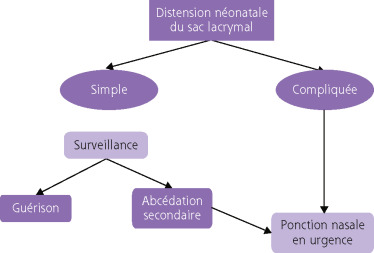
Fig. 7-13 Algorithme des choix thérapeutiques concernant les distensions néonatales du sac lacrymal.
L’anomalie diverticulaire peut exister dès le jeune âge, mais ne se révéler qu’à l’âge adulte.
Cliniquement, le maximum de la voussure se situe certes en dessous du tendon canthal médial mais volontiers latéral dans la paupière. Lors de la pression digitale, le reflux de mucus par les canalicules est inconstant, l’évacuation pouvant se faire directement dans le sac, puis par le canal lacrymonasal. Le contact osseux est présent.
Évolution. Les diverticules peuvent rester quiescents et n’être que peu gênants sauf s’ils sont associés à une imperforation lacrymonasale. Ils peuvent se surinfecter (parfois un mode révélateur). La diverticulite aiguë ressemble à un abcès du sac lacrymal. Ils peuvent redevenir quiescents à la disparition de l’infection. La prévention des récidives est chirurgicale : excision en bloc jusqu’à la face externe du sac. La dissection à chaud est difficile; retrouver le cordon à froid l’est aussi mais différemment.
Tout peut se rencontrer. On pensera d’autant plus à une lésion extralacrymale que la clinique est dépourvue d’épiphora et que la lésion n’est pas strictement localisée en regard de la gouttière lacrymale. L’imagerie est d’un grand secours.
Conflits d’intérêts : B. Fayet à un intérêt financier dans la commercialisation de la Masterka™.
[1] Soliman M, Lueder GT Initial management of congenital canalicular atresia J AAPOS 2015 ; 19 : 220-222
[2] Lyon DB, Dortzbach RK, Lemke BN, Gonnering RS Canalicular stenosis following probing for congenital nasolacrimal duct obstruction Ophthalmic Surg 1991 ; 22 : 228-232
[3] Chen L, Fang J Proximal drainage plus massage of lacrimal sac improves the symptoms of congenital dacryocystoceles Eur J Ophthalmol 2015 ; 25 : 293-297
[4] Karti O, Karahan E, Acan D, Kusbeci T The natural process of congenital nasolacrimal duct obstruction and effect of lacrimal sac massage Int Ophthalmol 2016 ; 36 : 845-849
[5] Jones LE, Wobig J Surgery of the eyelid and lacrimal system Birmingham (Alabama): Aesculapius (1976).
[6] Mocan MC, Gulmez Sevim D, Kocabeyoglu S, Irkec M Prognostic value of metalmetal contact during nasolacrimal duct probing Can J Ophthalmol 2015 ; 50 : 314-317
[7] Alanon-Fernandez MA, Alanon-Fernandez FJ, Martinez-Fernandez A, et al. Comparative study of primary intention lacrimal probing with and without nasal endoscopy Acta Otorrinolaringol Esp 2014 ; 65 : 297-301
[8] Baggio E, Ruban JM, Sandon K Analysis of the efficacy of early probing in the treatment of symptomatic congenital lacrimal duct obstruction in infants. Apropos of 92 cases J Fr Ophtalmol 2000 ; 23 : 655-662
[9] Le Garrec J, Abadie-Koebele C, Parienti JJ, et al. Nasolacrimal duct office probing in children under the age of 12 months : cure rate and cost evaluation J Fr Ophtalmol 2016 ; 39 : 171-177
[10] Dehghani N, Fouladivanda MR, Ghobadifar MA, et al. Nine-month follow-up results of treatment for nasolacrimal duct obstruction by probing with adjunctive mitomycin C in adults : a prospective randomized placebo-controlled trial Chonnam Med J 2015 ; 51 : 19-25
[11] Miller AM, Chandler DL, Repka MX, et al. Office probing for treatment of nasolacrimal duct obstruction in infants J AAPOS 2014 ; 18 : 26-30
[12] Kushner BJ Early office-based vs late hospital-based nasolacrimal duct probing Arch Ophthalmol 1995 ; 113 : 1103-1104
[13] Fayet B, Katowitz WR, Racy E, et al. Pushed monocanalicular intubation : an alternative stenting system for the management of congenital nasolacrimal duct obstructions J AAPOS 2012 ; 16 : 468-472
[14] Lala-Gitteau E, Majzoub S, Pisella PJ Use of MEOPA during nasolacrimal duct probing in children J Fr Ophtalmol 2007 ; 30 : 924-927
[15] Pediatric Eye Disease Investigator Group Resolution of congenital nasolacrimal duct obstruction with nonsurgical management Arch Ophthalmol 2012 ; 130 : 730-734
[16] Nelson LB Common management of congenital nasolacrimal duct obstruction J Pediatr Ophthalmol Strabismus 2015 ; 52 : 12
[17] Andalib D, Nabei R Intraoperative prognostic factors for probing outcome in children with congenital nasolacrimal duct obstruction Eur J Ophthalmol 2013 ; 23 : 329-332
[18] Fujimoto M, Ogino K, Matsuyama H, Miyazaki C Success rates of dacryoendos-copy-guided probing for recalcitrant congenital nasolacrimal duct obstruction Jpn J Ophthalmol 2016 ; 60 : 274-279
[19] Pediatric Eye Disease Investigator Group, Repka MX, Chandler DL, Bremer DL, et al. Repeat probing for treatment of persistent nasolacrimal duct obstruction J AAPOS 2009 ; 13 : 306-307
[20] Okumus S, Oner V, Durucu C, et al. Nasolacrimal duct intubation in the treatment of congenital nasolacrimal duct obstruction in older children Eye (Lond) 2016 ; 30 : 85-88
[21] Lee KA, Chandler DL, Repka MX, et al. A comparison of treatment approaches for bilateral congenital nasolacrimal duct obstruction Am J Ophthalmol 2013 ; 156 : 1045-1050
[22] Goldstein SM, Goldstein JB, Katowitz JA Comparison of monocanalicular stenting and balloon dacryoplasty in secondary treatment of congenital nasolacrimal duct obstruction after failed primary probing Ophthal Plast Reconstr Surg 2004 ; 20 : 352-357
[23] Wesley RE Inferior turbinate fracture in the treatment of congenital nasolacrimal duct obstruction and congenital nasolacrimal duct anomaly Ophthalmic Surg 1985 ; 16 : 368-371
[24] Wagner RS Management of congenital nasolacrimal duct obstruction Pediatr Ann 2001 ; 30 : 481-488
[25] Schellini SA, Ariki CT, Sousa RL, et al. Management of congenital nasolacrimal duct obstruction–latin american study Ophthal Plast Reconstr Surg 2013 ; 29 : 389-392
[26] Dotan G, Nelson LB Congenital nasolacrimal duct obstruction : common management policies among pediatric ophthalmologists J Pediatr Ophthalmol Strabismus 2015 ; 52 : 14-19
[27] Al-Faky YH, Mousa A, Kalantan H, et al. A prospective, randomised comparison of probing versus bicanalicular silastic intubation for congenital nasolacrimal duct obstruction Br J Ophthalmol 2015 ; 99 : 246-250
[28] Katowitz JA, Welsh MG Timing of initial probing and irrigation in congenital nasolacrimal duct obstruction Ophthalmology 1987 ; 94 : 698-705
[29] Maheshwari R Success rate and cause of failure for late probing for congenital nasolacrimal duct obstruction J Pediatr Ophthalmol Strabismus 2008 ; 45 : 168-171
[30] Hayashi K, Katori N, Komatsu H, Ohno-Matsui K Spontaneous resolving rate of congenital nasolacrimal duct obstruction and success rate of late probing after age 18 months : historical cohort study Nihon Ganka Gakkai Zasshi 2014 ; 118 : 91-97
[31] Zilelioglu G, Hosal BM The results of late probing in congenital nasolacrimal duct obstruction Orbit 2007 ; 26 : 1-3
[32] el-Mansoury J, Calhoun JH, Nelson LB, Harley RD Results of late probing for congenital nasolacrimal duct obstruction Ophthalmology 1986 ; 93 : 1052-1054
[33] Nelson LB Late probing success for congenital nasolacrimal duct obstruction J Pediatr Ophthalmol Strabismus 2008 ; 45 : 138
[34] Abrishami M, Bagheri A, Salour SH, Mirdehghan SA Late probing for congenital nasolacrimal duct obstruction J Ophthalmic Vis Res 2009 ; 4 : 102-104
[35] Baran F, Kelly JP, Finn LS, et al. Evaluation and treatment of failed nasolacrimal duct probing in Down syndrome J AAPOS 2014 ; 18 : 226-231
[36] Al-Faky YH, Al-Sobaie N, Mousa A, et al. Evaluation of treatment modalities and prognostic factors in children with congenital nasolacrimal duct obstruction J AAPOS 2012 ; 16 : 53-57
[37] Denis D, Saracco JB, Triglia JM Nasolacrimal duct cysts in congenital dacryocystocele Graefes Arch Clin Exp Ophthalmol 1994 ; 232 : 252-254