Décollement de rétine
P. Dureau, G. Caputo, F. Metge-Galatoire
Les décollements de rétine (DR) de l’enfant constituent 5 à 12 % de tous les DR [1]. L’enjeu est important en termes de pronostic visuel car, compte tenu des étiologies, l’oeil adelphe est moins souvent normal et fonctionnel que chez l’adulte (malformations, décollements bilatéraux).
Comme chez l’adulte, on distingue les décollements rhegmatogènes, exsudatifs et tractionnels. Toutefois, la répartition des différentes étiologies n’est pas la même : les décollements traumatiques, généralement rhegmatogènes, dominent (jusqu’à 50 % ). Il en découle une prédominance masculine marquée (jusqu’à 80 % dans certaines séries). Par ailleurs, les malformations congénitales constituent une étiologie spécifique à l’enfant, de prise en charge difficile en raison des particularités chirurgicales, des anomalies générales souvent associées, de la fréquente bilatéralité et de l’amblyopie.
Le retard diagnostique est fréquent chez l’enfant, entraînant une incidence supérieure de prolifération vitréorétinienne. Enfin, l’évolution vers des complications sévères pouvant aller jusqu’à la phtyse est possible et d’autant plus rapide que l’enfant est jeune.
Les signes d’appel sont variables mais ont en commun le retard fréquent par rapport à l’apparition du DR :
- baisse d’acuité visuelle découverte à l’occasion d’un examen systématique, d’un dépistage scolaire ;
- strabisme traduisant la mauvaise vision unilatérale ;
- leucocorie tardive liée au décollement total et/ou à une cataracte associée ;
- pathologie oculaire et/ou générale associée à un DR ;
- contexte familial ;
- contexte traumatique, pas toujours rapporté par l’enfant.
L’interrogatoire précise l’ancienneté des signes, les antécédents personnels (grossesse, accouchement, pathologies générales, malformations, traumatismes éventuels) et familiaux. Dans nombre de cas, l’examen de la famille peut apporter des éléments utiles au diagnostic.
La mesure de l’acuité visuelle est à adapter à l’âge de l’enfant. Chez les plus petits, une fixation stable et une bonne poursuite (en monoculaire) sont des éléments positifs. L’examen du segment antérieur (en position allongée sur le ventre face à la lampe à fente pour les nourrissons) peut permettre de noter une chambre antérieure étroite, une microphtalmie (surtout dans les pathologies malformatives), une rubéose irienne ou une cataracte. L’examen du segment postérieur est plus facile à l’ophtalmoscope indirect sur un enfant allongé, éventuellement avec une lentille de 28 D. Les éléments habituels sont notés : vitré (présence de sang, de signes d’inflammation, de malformations), rétine (topographie du décollement, déchirures, vaisseaux, état maculaire, prolifération vitréorétinienne). Toutefois, un examen complet, en particulier de la périphérie, n’est pas toujours possible. Dans ces cas, un examen sous anesthésie générale, souvent préopératoire immédiat, est nécessaire.
Les examens complémentaires sont mis en oeuvre soit lors de l’examen en consultation, soit lors de l’examen sous anesthésie générale :
- les rétinophotographies donnent parfois plus de renseignements que l’examen clinique chez un enfant qui bouge beaucoup, en raison de la brièveté du temps d’acquisition ;
- l’angiographie et les clichés en autofluorescence sont utiles, en particulier dans les pathologies vasculaires ;
- les systèmes d’imagerie grand champ (Optomap® et surtout RetCam™) donnent des images de toute la rétine ;
- la tomographie par cohérence optique (optical coherence tomography [OCT]) existe sous forme portable utilisable sous anesthésie générale ;
- l’échographie, souvent couplée au Doppler, est un examen clé dans les pathologies rétiniennes de l’enfant (fig. 19-1). Elle permet en cas de trouble des milieux de diagnostiquer : le décollement et ses caractéristiques de topographie, de mobilité, de vascularisation ; les déchirures ; l’état du vitré ; les lésions associées, etc. Son rôle pour éliminer un rétinoblastome, toujours redouté dans les pathologies du segment postérieur de l’enfant, est essentiel ;
- l’imagerie par résonance magnétique (IRM), nécessitant souvent une anesthésie générale chez l’enfant, est surtout utile en cas de tumeur ou de lésion associée ;
- la tomodensitométrie est surtout utile dans les contextes traumatiques (corps étrangers, os).
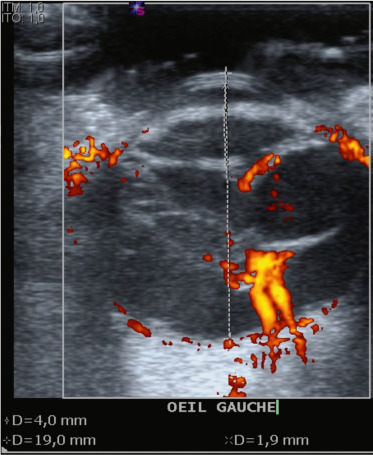
Fig. 19-1 Échographie : le Doppler permet d’objectiver les vaisseaux de la rétine décollée.
(Remerciements au Dr O. Bergès.)
Voir chapitre 25.2.
Les colobomes correspondent à un défaut de fermeture de la fente embryonnaire dans les premières semaines de la vie intra-utérine. Ce mécanisme explique la localisation habituellement nasale inférieure de la malformation. La forme la plus typique est choriorétinienne, impliquant la papille et une zone plus ou moins étendue de rétine adjacente. Il existe à la surface du colobome une membrane dite intercalaire qui correspond au prolongement de la rétine interne [2]. Avec l’âge, cette membrane s’atrophie et peut devenir le siège de trous souvent localisés près des bords du colobome. Ces trous livrent passage à du liquide et sont à l’origine du décollement (fig. 19-2). La prévalence de ce DR sur colobome choriorétinien est de 8 à 40 % selon les séries [3].
Le traitement chirurgical du DR sur colobome choriorétinien de l’enfant est difficile [2]. Les indentations ont un intérêt limité en raison de la difficulté de localisation des déhiscences dans la membrane intercalaire fine en regard de la sclère. La vitrectomie est la technique de choix. Le tamponnement comporte un risque de passage du silicone dans l’espace creux du colobome. La rétinopexie est assurée par du laser sur les bords du colobome.
Un cas particulier est celui des fossettes colobomateuses de la papille. Dans ces cas, le mécanisme d’apparition d’un décollement maculaire n’est pas complètement élucidé. Il pourrait s’agir d’une traction vitréenne sur les bords de la fossette entraînant, par une déhiscence, le passage de liquide sous la rétine en provenance du vitré liquéfié (fig. 19-3). Ce décollement avec baisse d’acuité visuelle est bien objectivé par l’OCT. Le traitement associe diversement vitrectomie, tamponnement par gaz et endophotocoagulation sur le bord de la papille.
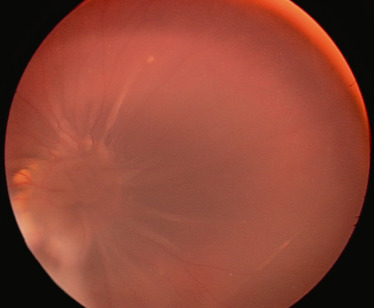
Fig. 19-2 Décollement de rétine total dans un cas de colobome, que l’on distingue dans la localisation habituelle en nasal inférieur de la papille.
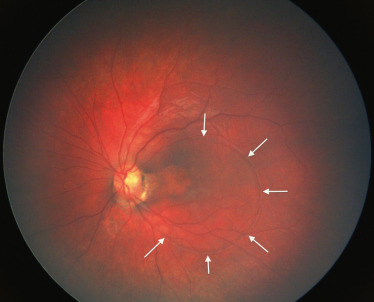
Fig. 19-3 Décollement maculaire (flèches) à partir d’une fossette colobomateuse de la papille.
Après chirurgie de cataracte congénitale, un décollement peut survenir plusieurs décennies après l’intervention (30 ans en moyenne dans une série de 52 cas en 2005 [4]). Comme chez l’adulte, le mécanisme est souvent initialement tractionnel avec la présence de vitré déplacé vers l’avant lors de l’intervention, entraînant des déhiscences périphériques en fer à cheval, plus rarement des trous atrophiques, des dialyses à l’ora ou des déchirures géantes. La nécessité d’ouvrir la capsule postérieure lors des interventions de cataracte chez les plus jeunes enfants favorise ces tractions vitréorétiniennes, surtout si une mèche de vitré reste incarcérée dans le segment antérieur, ce qui doit absolument être évité. Il est probable que l’évolution des techniques chirurgicales pour la cataracte de l’enfant (ablation de toutes les masses résiduelles, vitrectomie) fera diminuer l’incidence des DR postopératoires. Actuellement, les cas qui se compliquent de décollement, au moins précoce, sont majoritairement ceux dont la chirurgie a été difficile (hémorragies, malformations, etc.).
Toutefois, il persiste des difficultés spécifiques à l’enfant dans la prise en charge de ces décollements : myosis, synéchies et opacification des résidus capsulaires fréquents rendant difficile la visualisation de la rétine périphérique, cohérence du vitré qui n’est pas toujours détaché. La tendance est de privilégier les techniques de chirurgie endoculaire avec système de visualisation grand champ, éventuellement associées à un cerclage. Le pronostic reste médiocre en raison du retard diagnostique, de la prolifération vitréorétinienne fréquente et de l’amblyopie.
Après chirurgie d’ectopie du cristallin, un décollement peut également survenir. L’ectopie est liée dans plus de la moitié des cas à une maladie de Marfan. Dans cette pathologie, la mutation sur le gène de la fibrilline entraîne une fragilité zonulaire expliquant l’ectopie, mais aussi des anomalies de la sclère, qui est anormalement fine, et du vitré. Celui-ci est précocement liquéfié avec des zones d’adhérence anormale à la rétine. La sclère amincie se distend avec apparition d’une myopie axile. Les tractions vitréennes sont responsables de déhiscences rétiniennes. Enfin, l’ablation du cristallin fréquemment rendue nécessaire par l’ectopie et les troubles réfractifs qui en découlent est un facteur de risque supplémentaire en raison de la manipulation peropératoire du vitré. Ainsi, le risque de DR en cas d’aphaquie dans le cadre d’une maladie de Marfan atteint 30 % dans certaines séries [5]. Pour la même raison, les cas bilatéraux ne sont pas rares. L’utilisation de techniques complexes d’implant suturé peut favoriser les complications et rendre la chirurgie du décollement plus difficile. Le traitement chirurgical se rapproche de celui des décollements après chirurgie de cataracte congénitale.
La persistance de la vascularisation foetale (PVF) peut s’accompagner de DR faisant partie de la malformation initiale ou postopératoire. La forme typique de cette malformation unilatérale comporte une microphtalmie avec une opacité blanche rétrocristallinienne vascularisée et un étirement des procès ciliaires, ainsi qu’un reliquat plus ou moins important d’artère hyaloïde reliant la papille à la face postérieure du cristallin. Il existe par ailleurs des formes postérieures (isolées ou plus souvent associées à la forme antérieure). Ces formes associent diversement des tractions de reliquats du système hyaloïdien sur la rétine et le cristallin et des décollements plus ou moins étendus (fig. 19-4) [6]. La croissance du globe, alors que les reliquats fibreux sont inextensibles, augmente les tractions et aggrave les décollements. Par ailleurs, lors d’une intervention sur la malformation antérieure, les anomalies d’insertion antérieure de la rétine (absence de pars plana) exposent au risque de déhiscence iatrogène. Ainsi, une intervention pour ces malformations est généralement indiquée, même si le pronostic fonctionnel des formes mixtes ou postérieures est médiocre, en raison du risque d’aggravation de la situation dans les premières années de vie. Une vitrectomie avec coagulation des résidus vasculaires et section des tractions est généralement pratiquée [7].
Après chirurgie de glaucome congénital, un décollement peut survenir en raison de la myopie forte induite par la buphtalmie et des complications de la chirurgie (hémorragie intravitréenne, décollement choroïdien, issue de vitré) (fig. 19-5). La prévalence de ces décollements a été estimée à 4 % [8]. Parmi les enfants opérés de glaucome congénital, 15 % présentent des lésions rétiniennes périphériques (palissades, déchirures, trous) qui devraient être systématiquement recherchées. Le délai entre l’intervention de glaucome et le décollement peut être de plusieurs années. Le pronostic fonctionnel réservé du décollement s’additionne à celui du glaucome.
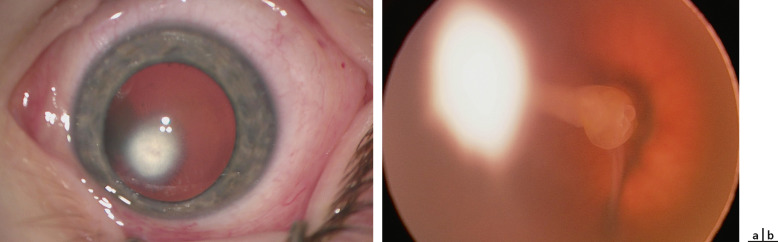
Fig. 19-4 Persistance de la vascularisation foetale : opacité blanche rétrocristallinienne décalée en nasal (a) reliée à un décollement venant de la papille (b).
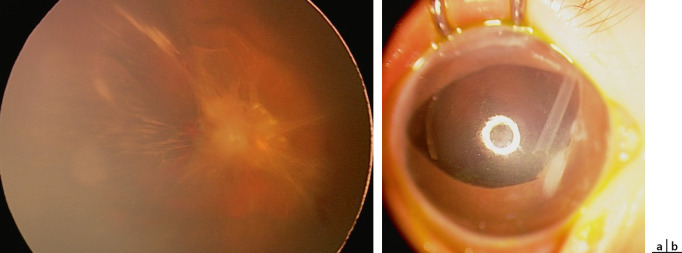
Fig. 19-5 Décollement postopératoire (a) chez un enfant traité pour glaucome congénital par une valve de dérivation (b).
La myopie forte constitue un quart à un tiers des étiologies de DR de l’enfant (les traumatismes représentant quant à eux environ la moitié des étiologies) [9]. Parmi les enfants ayant une myopie supérieure à −6 D, environ un tiers présente des lésions périphériques : palissades, blanc sans pression, trous [10]. Le caractère cohérent et souvent non décollé du vitré des enfants augmente le risque de décollement secondaire à ces lésions. Il n’existe pas de consensus sur un traitement préventif des lésions périphériques, d’autant que ce traitement peut être difficile à mettre en oeuvre sans anesthésie générale si l’enfant est petit. Les DR secondaires à une myopie forte chez l’enfant, comme tous les décollements sur ce terrain, se caractérisent souvent par un retard au diagnostic et donc une importante prolifération vitréorétinienne. Le traitement par cerclage peut être efficace en première intention dans ces yeux fragiles [11], mais une vitrectomie est souvent nécessaire, en particulier si la myopie est supérieure à −10 D [12].
Le syndrome de Stickler est une vitréorétinopathie qui associe à une myopie forte des anomalies faciales (aplatissement médiofacial), ORL (fente palatine, syndrome de Pierre-Robin avec rétrognathisme et glossoptose, surdité), dentaires, articulaires. La transmission est autosomique dominante avec des mutations des gènes COL2A1 ou COL11A1 codant pour le collagène. On distingue le type 1 où le vitré est le siège de voiles (fig. 19-6), et le type 2 où il comporte des condensations fibrillaires. La myopie est généralement forte, précoce et non évolutive. Une cataracte peut apparaître. Le risque de DR est très élevé (jusqu’à 50 % ), avec souvent des déchirures géantes. Le traitement associe généralement un cerclage à une chirurgie ab interno. Certaines équipes proposent un traitement préventif par laser ou cryo-application [13].
Le syndrome de Wagner et la vitréorétinopathie érosive, purement oculaires, se rapprochent du syndrome de Stickler avec toutefois moins de DR.
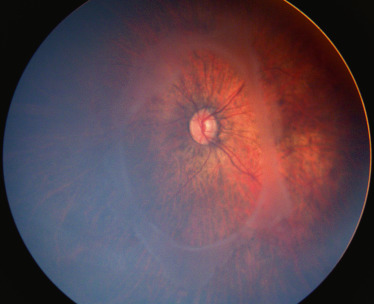
Fig. 19-6 Syndrome de Stickler : myopie forte, voiles vitréens.
Le rétinoschisis juvénile lié à l’X (voir chapitre 15) se caractérise par un clivage dans la couche plexiforme externe au niveau fovéolaire, responsable de la baisse de vision à partir de l’âge scolaire, et au niveau périphérique (fig. 19-7). Ces poches de schisis périphériques, parfois très saillantes, peuvent se compliquer d’hémorragies, par rupture du feuillet interne au niveau d’un vaisseau, et de DR. Le décollement peut être rhegmatogène par déhiscence dans le feuillet externe ou tractionnel par adhérence entre le vitré et le feuillet interne [14]. En cas de décollement tractionnel, une chirurgie externe peut être réalisée, si la déhiscence dans le feuillet externe est repérable, ce qui n’est pas toujours facile. Dans les autres cas ou si le décollement est tractionnel, une vitrectomie avec tamponnement est nécessaire. Une rétinectomie du feuillet interne est souvent inévitable dans ces cas. À noter qu’après vitrectomie, des régressions du schisis maculaire ont été décrites, vraisemblablement en supprimant un mécanisme tractionnel [15].
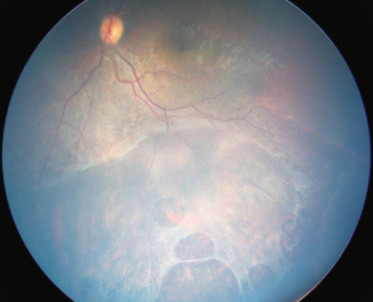
Fig. 19-7 Rétinoschisis juvénile lié à l’X : schisis périphérique avec déhiscences dans le feuillet externe.
Le syndrome d’Ehlers-Danlos est une maladie autosomique dominante du tissu collagène caractérisée par une hyperextensibilité cutanée et ligamentaire et des anévrismes aortiques. Elle peut se compliquer d’ectopie du cristallin, de stries angioïdes, de fragilités sclérales et d’anomalies vitréorétiniennes : liquéfaction précoce du vitré, tractions et hémorragies vitréennes, déchirures pouvant se compliquer de décollement.
La dysplasie de Kniest est une affection du collagène de type 2 qui comporte petite taille, arthropathie, fente palatine et surdité.
Les complications oculaires sont similaires à celles du syndrome de Stickler : myopie forte, anomalies vitréennes, déchirures géantes et décollement rhegmatogène.
La dysplasie spondylo-épiphysaire, autosomique dominante, est également une affection du collagène de type 2. La petite taille s’associe à une scoliose et des membres brefs. La myopie forte peut se compliquer de déchirures géantes et de décollement.
Une dialyse à l’ora idiopathique peut survenir chez l’enfant et se compliquer de DR [16]. La localisation est généralement temporale inférieure, parfois bilatérale. Les décollements sont à progression lente avec des lignes de démarcation, des kystes rétiniens, des zones de dégénérescence microkystique. La prolifération vitréorétinienne est rare. Le traitement préférentiel est la photocoagulation dans les formes non décollées, la cryo-indentation pour les décollements.
La prévention des décollements rhegmatogènes chez l’enfant n’a pas fait l’objet d’études permettant de prendre une décision sur des bases irréfutables, en raison de : la rareté des cas ; la pauvreté des symptômes rapportés par l’enfant ; la difficulté d’examen de la périphérie rétinienne ; la multiplicité des étiologies ; etc.
Il existe quelques études faisant état d’une prévention efficace, par laser ou cryothérapie, dans les syndromes de Stickler [13]. Dans les autres cas, la décision dépend du chirurgien et des circonstances (découverte lors d’un examen sous anesthésie générale par exemple).
Voir chapitre 16.3.
Chez l’enfant, les hémangiomes choroïdiens peuvent, rarement, être isolés et localisés comme chez l’adulte, mais s’intègrent le plus souvent dans un syndrome de Sturge-Weber. Cette neurofibromatose qui se complique d’hémangiomes choroïdiens dans 30 à 50 % des cas se caractérise par la présence unilatérale d’angiomes cutanés, oculaires et du système nerveux central : angiome de l’hémiface dans le territoire du trijumeau et particulièrement du V1 dans les formes avec complications oculaires ; angiome méningé (pie-mère) responsable d’une épilepsie de traitement difficile et souvent d’un retard mental ; angiome épiscléral responsable d’un glaucome, également de traitement difficile ; angiome choroïdien diffus. Cet angiome qui occupe généralement la plus grande partie de la choroïde se caractérise cliniquement par une couleur plus rouge du fond d’oeil du côté intéressé, un aspect « succulent » de la rétine (vaisseaux choroïdiens peu visibles). L’examen complémentaire le plus utile est l’échographie qui montre un épaississement choroïdien diffus. L’apparition d’un décollement exsudatif est fréquente et, avec le glaucome, l’atteinte du système nerveux central conditionne le pronostic fonctionnel (fig. 19-8). Le traitement de ce décollement est difficile, la photothérapie dynamique n’étant applicable qu’aux formes localisées. La protonthérapie (20 Gy) permet un assèchement de l’angiome et la réapplication de la rétine, ainsi qu’une amélioration du glaucome associé [17].
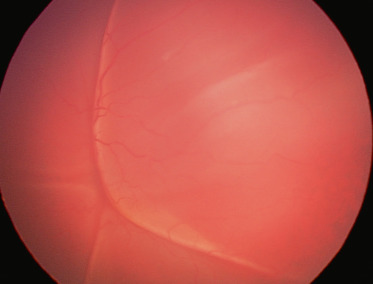
Fig. 19-8 Décollement exsudatif sur angiome choroïdien dans un syndrome de Sturge-Weber
Noter la couleur rouge de la choroïde sous-jacente.
Le DR exsudatif est une des formes caractéristiques de révélation du rétinoblastome (voir chapitre 20) et cette tumeur doit toujours être évoquée en premier. Après traitement conservateur, peuvent apparaître des décollements rhegmatogènes, en rapport avec des déhiscences au bord de la cicatrice, ou tractionnels en raison de brides vitréennes ou de séquelles d’hémorragie intravitréenne (fig. 19-9) [18].
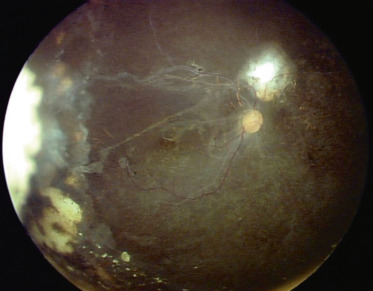
Fig. 19-9 Décollement tractionnel après traitement conservateur de rétinoblastome.
Après chirurgie filtrante chez l’enfant, en particulier après la pose d’une valve de drainage de l’humeur aqueuse, la chute brutale de pression intra-oculaire et la faible rigidité sclérale favorisent l’apparition d’un décollement choroïdien qui peut se compliquer de décollement exsudatif. Si le décollement se prolonge plus d’une semaine ou deux, souvent associé à une athalamie et une hypotonie prolongée avec filtration excessive, un drainage de l’hématome par voie sclérale est justifié. La réapplication rétinienne peut laisser des séquelles à type de remaniement pigmentaire, avec parfois une baisse définitive d’acuité visuelle par rapport à la période préopératoire. En raison du caractère cohérent et non décollé du vitré de l’enfant, la réapplication rétinienne comporte également un risque de décollement, tractionnel cette fois.
Les sclérites postérieures sont rares chez l’enfant, souvent idiopathiques, et peuvent se compliquer de DR exsudatif. Le traitement est celui de l’inflammation.
Le syndrome de Vogt-Koyanagi-Harada associe des signes généraux (cutanés, méningés, ORL) à des signes oculaires : panuvéite granulomateuse, oedème papillaire, décollement exsudatif. Le diagnostic est souvent retardé chez l’enfant [19]. Le traitement est celui de l’inflammation (voir chapitre 14).
Voir chapitre 16.1.
Voir chapitre 15.
Voir chapitre 25.2.
En raison du caractère cohérent et non décollé du vitré de l’enfant, une hémorragie intravitréenne peut se compliquer rapidement (quelques jours ou semaines) de fibrose et de tractions responsables d’un décollement. Ces hémorragies peuvent s’observer, en dehors des traumatismes, dans diverses pathologies oculaires et/ou générales :
- rétinoschisis juvénile lié à l’X, où les vaisseaux passant dans les zones de schisis peuvent se rompre et saigner dans la cavité schisique et le vitré. Ce mécanisme tractionnel peut se combiner à un décollement rhegmatogène en cas de déhiscence dans le feuillet externe ;
- vitréorétinopathie exsudative familiale où des néovaisseaux peuvent saigner ;
- rétinopathie des prématurés avec des néovaisseaux à la limite de la rétine non vascularisée, responsables d’hémorragies et de tractions (stade 4 puis 5) ;
- maladie de Coats : saignements à partir des anomalies vasculaires (télangiectasies) ;
- uvéites intermédiaires et postérieures compliquées de néovaisseaux ;
- malformations vasculaires (anévrismes, angiomes, boucles, télangiectasies, communications artérioveineuses, etc.) ;
- drépanocytose avec ischémie rétinienne périphérique, néovaisseaux et saignement ;
- causes hématologiques de saignement (leucémies, thrombopénie, hémophilie, déficit en protéine C, maladie de von Willebrandt, etc.).
Cette affection dominante liée à l’X touche presque uniquement les filles, étant létale chez les garçons. Elle est liée à une mutation du gène NEMO (NFκB Essential MOdulator). Elle se traduit par une éruption cutanée linéaire bulleuse des membres dans les premières semaines de vie. Cette éruption disparaît mais peuvent apparaître, dans les premiers mois de vie, des anomalies rétiniennes périphériques : tortuosité vasculaire, shunts, zones avasculaires [20]. Ces anomalies généralement unilatérales et temporales évoluent de façon comparable à la rétinopathie des prématurés : non-perfusion, apparition de néovaisseaux, saignements, tractions et DR (fig. 19-10). Un traitement par photocoagulation peut stopper cette évolution. Le caractère précoce exige une surveillance rapprochée du fond d’oeil, tous les mois pendant les 3 premiers mois de vie puis de façon plus espacée.
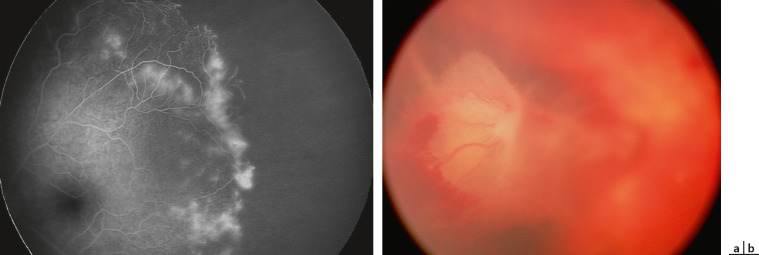
Fig. 19-10 Incontinentia pigmenti
a. Zones de non-perfusion et néovaisseaux périphériques. b. Décollement tractionnel hémorragique.
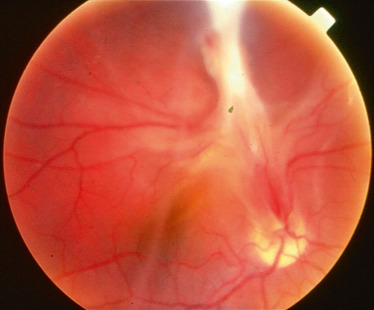
Lorsqu’il existe une inflammation du vitré dans un contexte infectieux ou plus souvent d’uvéite, les tractions générées peuvent être à l’origine d’un DR. La toxocarose par exemple est une source de hyalite souvent majeure (fig. 19-11). Dans ce contexte de décollement avec un engainement inflammatoire du corps ciliaire, le risque d’hypotonie et de phtyse est élevé. Le traitement repose sur la vitrectomie et le cerclage chirurgical qui lève les tractions.
[1] Caputo G. Indications chirurgicales et particularités techniques chez l’enfant. In : Caputo G, Metge-Galatoire F, Arndt C, Conrath J. Décollements de rétine, rapport SFO 2011. Issy-les-Moulineaux : Elsevier Masson ; 2011, p. 441-6.
[2] Teoh SCB, Mayer EJ, Haynes RJ, et al. Vitreoretinal surgery for retinal detachment in retinochoroidal colobomata. Eur J Ophthalmol 2008 ; 18 : 304-8.
[3] Unlü N, Kocaoğlan H, Acar MA, et al S. Surgical management of retinal detachment with choroidal coloboma. Eur J Ophthalmol 2002 ; 12 : 299-303.
[4] Yorston D, Yang YF, Sullivan PM. Retinal detachment following surgery for congenital cataract : presentation and outcomes. Eye 2005 ; 19 : 317-21.
[5] Chandra A, Ekwalla V, Child A, Charteris D. Prevalence of ectopia lentis and retinal detachment in Marfan syndrome. Acta Ophthalmol 2014 ; 92 : e82-83.
[6] Hu A, Pei X, Ding X, et al. Combined persistent fetal vasculature : a classification based on high-resolution B-mode ultrasound and color Doppler imaging. Ophthalmology 2016 ; 123 : 19-25.
[7] Bosjolie A, Ferrone P. Visual outcome in early vitrectomy for posterior persistent fetal vasculature associated with traction retinal detachment. Retina 2015 ; 35 : 570-6.
[8] Gupta S, Gogia V, Jose C, et al. Peripheral retinal degeneration and rhegmatogenous detachment in primary congenital glaucoma. Retina 2016 ; 36 : 188-91.
[9] Lee RWJ, Mayer EJ, Markham RH. The aetiology of paediatric rhegmatogenous retinal detachment : 15 years experience. Eye 2008 ; 22 : 636-40.
[10] Bansal AS, Hubbard GB. Peripheral retinal findings in highly myopic children < or = 10 years of age. Retina 2010 ; 30 : S15-19.
[11] Errera MH, Liyanage SE, Moya R, et al. Primary scleral buckling for pediatric rhegmatogenous retinal detachment. Retina 2015 ; 35 : 1441-9.
[12] Wang NK, Chen YP, Lai CC, et al. Paediatric retinal detachment : comparison of high myopia and extreme myopia. Br J Ophthalmol 2009 ; 93 : 650-5.
[13] Fincham GS, Pasea L, Carroll C, et al. Prevention of retinal detachment in Stickler syndrome: the Cambridge prophylactic cryotherapy protocol. Ophthalmology 2014 ; 121 : 1588-97.
[14] Guez-Daudin A. Décollement de rétine sur rétinoschisis juvénile lié à l’X. In : Caputo G, Metge-Galatoire F, Arndt C, Conrath J. Décollements de rétine, rapport SFO 2011. Issy-les-Moulineaux : Elsevier Masson ; 2011, p. 336-40.
[15] Ikeda F, Iida T, Kishi S. Resolution of retinoschisis after vitreous surgery in X-linked retinoschisis. Ophthalmology 2008 ; 115 : 718-722.e1.
[16] Snead MP. Retinal detachment in childhood. In : Taylor D, Hoyt CS. Pediatric ophthalmology and strabismus. London : Elsevier ; 2005, p. 595-605.
[17] Zografos L, Egger E, Bercher L, et al. Proton beam irradiation of choroidal hemangiomas. Am J Ophthalmol 1998 ; 126 : 261-8.
[18] Lumbroso-Le Rouic L. Décollement de rétine et rétinoblastome. In : Caputo G, Metge-Galatoire F, Arndt C, Conrath J. Décollements de rétine, rapport SFO 2011. Issyles- Moulineaux : Elsevier Masson ; 2011, p. 520-3.
[19] Daudin JB, Brezin AP. Décollements de rétine exsudatifs d’origine inflammatoire ou infectieuse. In : Caputo G, Metge-Galatoire F, Arndt C, Conrath J. Décollements de rétine, rapport SFO 2011. Issy-les-Moulineaux : Elsevier Masson ; 2011, p. 403-13.
[20] Chen CJ, Han IC, Goldberg MF. Variable expression of retinopathy in a pedigree of patients with incontinentia pigmenti. Retina 2015 ; 35 : 2627-32.