Les pathologies
Pathologie oculo-orbitaire
Coordonné par D. Denis
D. Denis, G. Pech-Gourg, P. Wary, D. Scavarda
Les craniosténoses sont des déformations crâniennes dues à la fermeture prématurée (synostose) (fig. 6-1) d’une ou de plusieurs sutures crâniennes qui surviennent approximativement dans 1/2 500 naissances [1]. La malformation osseuse congénitale s’oppose au développement cérébral et ce conflit a deux conséquences – l’une morphologique anatomique et l’autre cérébrale et ophtalmologique –, expliquant que les paramètres à évaluer vont devoir être :
la ou les sutures synostosées permettant une classification ;
les complications directes ou indirectes, neurologiques (hypertension intracrânienne [HTIC], retard mental) et ophtalmologiques (neuropathie optique, troubles réfractifs, strabisme, exophtalmieexorbitisme, souffrance cornéenne).
La physiopathogénie est inconnue malgré les avancées génétiques.
Les craniosténoses se caractérisent par une hétérogénéité génétique.Le rôle d’un âge paternel élevé est évoqué dans la survenue de mutations de novo. À ce jour, les mutations ont été retrouvées dans sept gènes différents : les gènes du récepteur FGFR1, FGFR2, FGFR3, les gènes MCX2, TWIST1, EFNB1 et EFNB2. Ces sept gènes sont impliqués dans 30 % des craniosténoses syndromiques [1] qui ont une transmission autosomique dominante à pénétrance variable.
L’étiologie génétique des formes isolées non syndromiques est encore mal connue. Les facteurs environnementaux sont évoqués mais mal documentés (tabac, contraintes intra-utérines, etc.) et pourraient expliquer les variations d’incidence des différents types [2].
La synostose prématurée d’une suture crânienne est un phénomène dynamique qui commence pendant la vie intra-utérine et se poursuit après la naissance : il y a arrêt de la croissance dans la direction perpendiculaire à la suture synostosée avec une croissance compen- satrice dans la direction parallèle à la suture atteinte (fig. 6-1) (loi de Virchow = allongement du diamètre parallèle à la suture sténosée).
À chaque suture crânienne concernée par le processus sténotique correspond un morphotype de craniosténose dont l’analyse permet une classification. La gravité de la craniosténose est fonction du nombre et du type de sutures concernées avec dans les formes sévères des complications neurologiques et ophtalmologiques (tableaux 6-1 et 6-2). Par ordre de fréquence décroissante [3], on retrouve la scaphocéphalie, la trigonocéphalie, la plagiocéphalie, devant la brachycéphalie, l’oxycéphalie puis les formes syndromiques.
La classification se fonde sur le contexte isolé ou syndromique et sur la ou les sutures synostosées (nombre) : la craniosténose étant uni- ou multisuturaire (fig. 6-2).
Les différentes craniosténoses sont détaillées dans le Tableau 6-1 selon le siège de la suture atteinte, la déformation crânienne et les signes associés (fig. 6-3 à fig. 6-7).
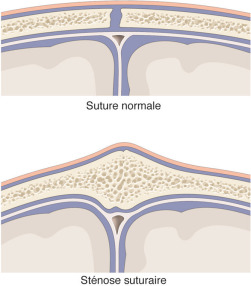
Fig. 6-1 Schémas histologiques d’une suture normale et d’une suture synostosée.
Tableau 6-1 - Classifi cation des différentes craniosténoses isolées : siège de la suture atteinte, déformation crânienne et signes ophtalmologiques.
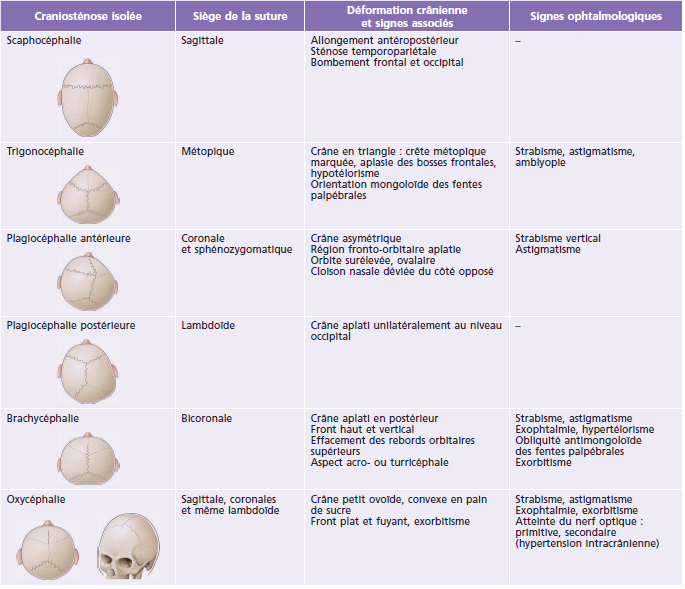
Tableau 6-2 – Craniosténoses syndromiques : siège de la suture atteinte, déformation crânienne et signes ophtalmologiques.
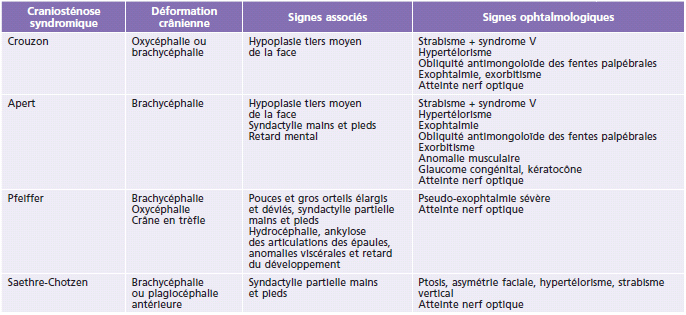
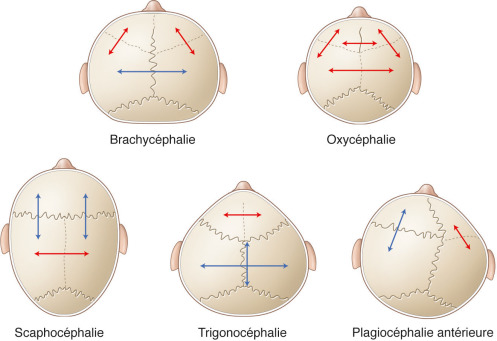
Fig. 6-2 Déformation crânienne due à un arrêt de la croissance osseuse dans la direction perpendiculaire à la suture synostosée et croissance compensatrice dans la direction parallèle.
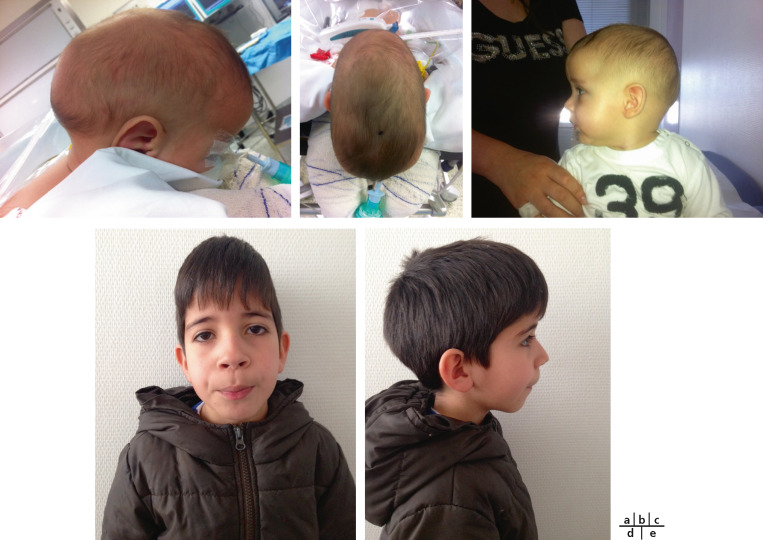
Fig. 6-3 Scaphocéphalie.
a. Profil : allongement dans le sens antéropostérieur avec bosses frontales proéminentes. b. Vue supérieure. c. Vue de profil après chirurgie correctrice. d. Scaphocéphalie découverte tardivement non opérée : vue de face, crâne haut et étroit. e. Scaphocéphalie découverte tardivement non opérée : vue de profil, allongement dans le sens antéropostérieur.
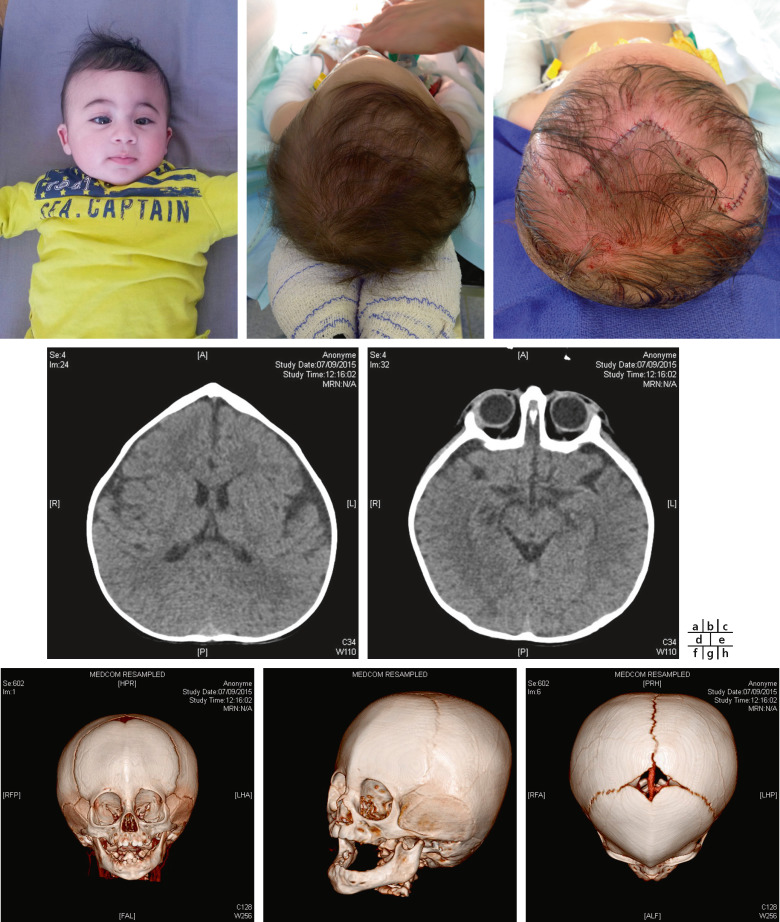
Fig. 6-4 Trigonocéphalie.
a. Vue de face : hypotélorisme ; épicanthus et sourcil en accent circonflexe. b. Vue supérieure préopératoire. c. Vue supérieure postopératoir. d. Scanner. e. Coupe axiale au niveau frontal montrant l’aspect caractéristique en carène de navire. f. Coupe axiale au niveau orbitaire (trigonaxial) montrant l’hypotélorisme caractéristique. g. Reconstruction tridimensionnelle de face et de trois quarts objectivant la fermeture de la suture métopique et l’hypotélorisme. h. Reconstruction tridimensionnelle en vue supérieure : bombement compensateur bilatéral au niveau de la région pariétale.
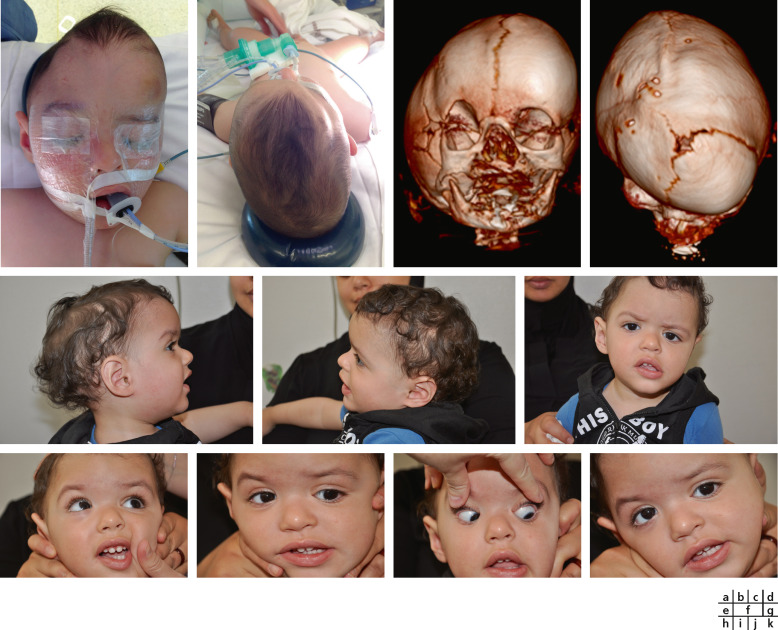
Fig. 6-5 Plagiocéphalie antérieure sévère : déviation de la face vers le côté dit « sain » .
a, b. Vues de face et supérieure objectivant l’aplatissement orbitaire supérieur droit, l’asymétrie faciale avec déviation de la cloison nasale vers le côté sain gauche et bombement frontal compensateur controlatéral droit. c, d. Reconstructions en 3D – vues antérieure et supérieure – montrant l’asymétrie orbitaire avec : déformation et ascension du toit de l’orbite en haut et en dehors, fermeture de la suture coronale droite qui est de ce fait invisible par rapport à la suture coronale gauche qui est normale ; recul de la bosse frontale droite. Il s’agit ici d’une forme complexe associée à une fermeture de la suture sagittale. e-k. En postopératoire : récupération du bombement frontal permettant une meilleure symétrie et par conséquent une esthétique très satisfaisante (e, f) ; persistance d’une scoliose faciale en rapport avec sa position de la tête : inclinaison tête sur épaule gauche (g). Paralysie du IV droit : hyperaction de l’oblique inférieur droit (h), hypoaction de l’oblique supérieur droit (i) et hyperaction du droit inférieur gauche (j) ; manoeuvre de Bielschowski : accentuation de l’élévation de l’oeil droit dans l’inclinaison forcée de la tête à droite (k).

Fig. 6-6 Évolution d’une plagiocéphalie opérée à l’âge de 6 mois sans prise en charge oculomotrice.
On note : de face (a), un aspect de « scoliose faciale » majeure avec torticolis sévère, tête penchée sur l’épaule gauche ; en vue supérieure (b), un remodelage fronto-orbitaire satisfaisant.
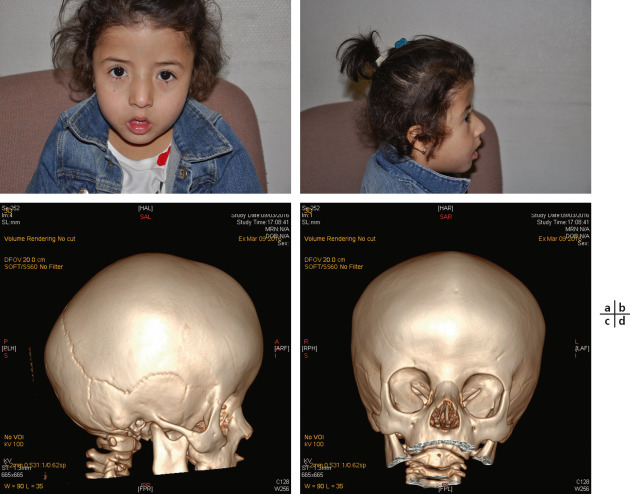
Fig. 6-7 Brachycéphalie non syndromique.
a. Vue de face : front ascensionné et élargi. b. Vue de profil : raccourcissement du diamètre antéropostérieur du crâne. c, d. Scanner en 3D, vues de face et de profil : raccourcissement de l’étage antérieur majeur avec bombement compensateur bilatéral au niveau de la région temporale.
Ce sont des formes cliniques complexes (Tableau 6-2) au pronostic neurologique ou fonctionnel plus sévère dont les plus fréquentes sont les syndromes décrits ci-dessous [3].
Incidence : 1/100 000 en France et 1/50 000 aux États-Unis.
Ce syndrome associe [4] une craniosténose de type brachycéphalie, une hypoplasie faciale qui occasionne des troubles de l’articulé dentaire, des problèmes respiratoires obstructifs, une obstruction lacrymonasale, un retard mental plus ou moins sévère (malformation cérébrale associée), des anomalies cardiaques et viscérales ainsi qu’une syndactylie des mains et des pieds (pouce et gros orteil) (fig. 6-8). L’inadéquation entre le contenu et la taille de l’orbite osseuse explique l’exorbitisme, moins sévère que dans le syndrome de Crouzon.
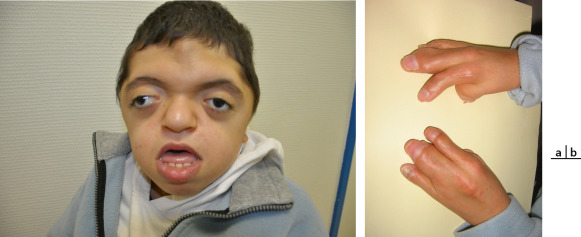
Fig. 6-8 Syndrome d’Apert.
a. Brachycéphalie et hypertélorisme, hypoplasie du massif facial moyen. b. Syndactylies des mains.
Incidence : 1/50 000 en France.
Syndrome caractérisé par une triade [5] : orbites courtes, craniosténose (type brachycéphalie ou oxycéphalie), hypoplasie maxillaire supérieure à laquelle s’ajoute un prognathisme relatif qui explique les troubles de l’articulé dentaire. Au niveau facial, l’hypoplasie osseuse résulte de la combinaison de facteurs qui affectent la base du crâne et la croissance de la voûte crânienne. L’étroitesse des orbites a pour conséquence un exorbitisme marqué avec pseudo-exophtalmie et hypertélorisme (fig. 6-9). Ce syndrome peut se compléter progressivement de la naissance jusqu’à l’âge de 2 à 3 ans environ. L’hypertension intracrânienne est la complication redoutée de ce syndrome.
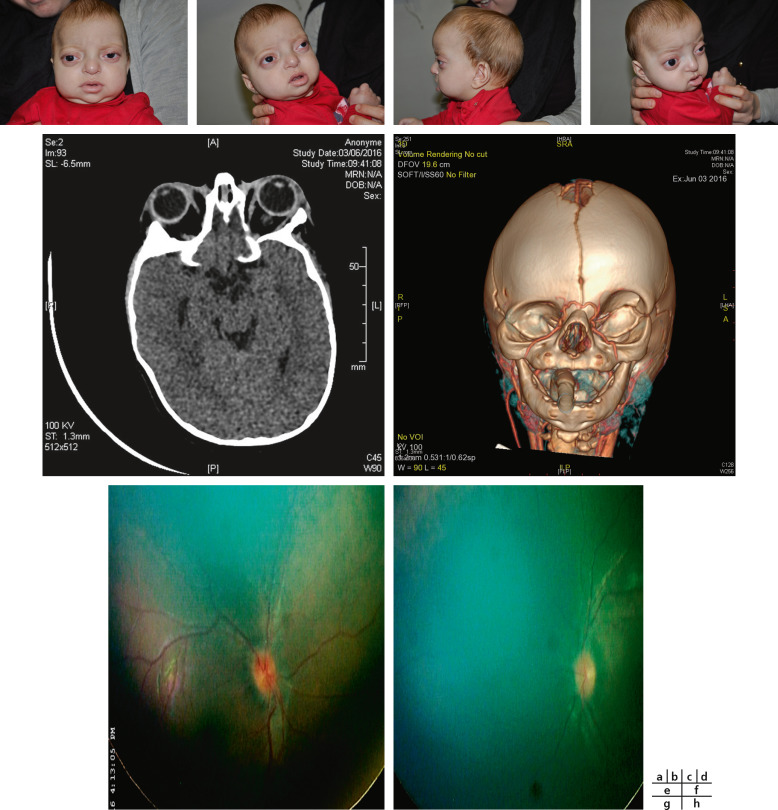
Fig. 6-9 Syndrome de Crouzon.
a. Vue de face, hypertélorisme, strabisme divergent et syndrome V. b. Divergence avec syndrome V. c. Vue de profil : hypoplasie faciale du tiers moyen de la face. d. Pseudo-exophtalmie. e. TDM en coupe axiale des orbites : extorsion, hypertélorisme important. f. Scanner 3D : aspect d’oxycéphalie (sutures sagittale, coronale et lambdoïde fermées) et impression digitiforme (reconstruction tridimensionnelle endocrânienne). g, h. OEdème papillaire (RetCam™ 3) diagnostiqué lors du bilan de syndrome de Crouzon imposant d’anticiper la prise en charge neurochirurgicale.
Incidence : 1/100 000 en France.
Craniosténose de type brachycéphalie, oxycéphalie ou crâne en trèfle avec hypoplasie faciale et anomalies des extrémités (pouces et gros orteils, syndactylie partielle) [6]. Les sutures fusionnées sont les sutures coronales et parfois sagittales. Une hydrocéphalie peut être parfois présente, ainsi qu’une pseudo-exophtalmie sévère, une ankylose des articulations des épaules, des anomalies viscérales et un retard du développement. Il existe trois types de syndrome de Pfeiffer selon l’expression clinique et la présence d’un retard mental. Le type 1 est le plus fréquent, où l’expression clinique est modérée et l’intelligence normale (encadré 6-1). Le type 2 associe retard mental, crâne en trèfle, pseudo-exophtalmie extrême, synostose des épaules et complications neurologiques. Le type 3 est analogue au type 2, mais sans crâne en trèfle (fig. 6-10).
Incidence : 1/50 000 en France.
Ce syndrome est une acro-céphalo-syndactylie qui présente une variabilité phénotypique importante [7]. La craniosténose secondaire à la soudure d’une hémi-coronale, rarement des deux coronales ou de la métopique, se traduit par une dysmorphie faciale (asymétrie faciale) avec hypertélorisme, une obliquité antimongoloïde et un ptosis. Une syndactylie des 4e et 5e doigts, des anomalies auditives, des anomalies des vertèbres cervicales et des malformations cardiaques congénitales sont associées. Une hypertension intracrânienne peut survenir.
Ces différentes craniosténoses simples et syndromiques provoquent des perturbations sévères craniofaciales au niveau de la voûte et de la base cartilagineuse du crâne, du massif maxillaire et des orbites. Ces perturbations de la croissance osseuse sont à l’origine d’un conflit avec les développements respectifs du cerveau et de l’oeil qui sont très importants jusqu’à l’âge de 3 ans, avec sur le plan oculaire l’apparition de la vision binoculaire (4,5 mois), de l’oculomotricité et de la fonction monoculaire (jusqu’à 10 ans). Les conséquences seront neurologiques, ophtalmologiques respiratoires et esthétiques et pourront menacer le pronostic cérébral et visuel.
Cas clinique : syndrome de Pfeiffer
Enfant adressé à l’âge de 2 ans pour exophtalmie bilatérale dans le cadre d’une faciocraniosténose syndromique de type Pfeiffer de type 1.
AV : 6/10 ODG R8. Réfraction : OD + 1,75 (–1,75 à 10°) ; OG + 2,25 (–1,25 à 150°).
Oculomotricité : XtOG 4°.
FO : oedème papillaire bilatéral +++++.
Après chirurgie : chute de l’AV ODG ; 3/10 OD ; VBLM OG (3 ans), mais disparition de l’oedème papillaire ; FO normal.
Traitement de l’amblyopie : 1er temps : occlusion 2 heures de l’OD pendant 4 semaines, permettant une remontée de l’AV à 10/10 OD et 1/10 OG. 2e temps : occlusion totale OD pendant 3 semaines permettant d’obtenir → 10/10 OD et 8/10 OG.
CONCLUSION : la surveillance ophtalmologique rapprochée avec traitement de l’amblyopie est indispensable en association à la chirurgie craniofaciale, permettant la récupération quasi totale de l’acuité visuelle ++++++.
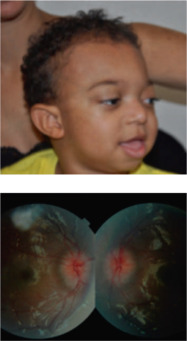
Avant chirurgie craniofaciale
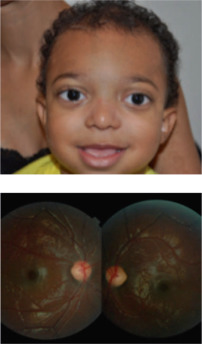
Après chirurgie craniofaciale
AV : acuité visuelle ; FO : fond d’oeil ; OD : oeil droit ; ODG : oeil droit et gauche ; OG : oeil gauche.
Fig. 6-10 Syndrome de Pfeiffer : brachycéphalie et hypoplasie du massif facial ; pseudo-exophtalmie et strabisme divergent chez un nourrisson de 5 mois.
Le diagnostic de craniosténose est clinique et doit être le plus précoce possible. Il peut être suspecté dans les premiers jours de vie. La déformation du crâne doit faire penser à une craniosténose. L’évolutivité de la déformation dans les mois qui suivent la suspicion du diagnostic impose un suivi rigoureux. Un bilan génétique est effectué quand une forme syndromique est suspectée.
Le diagnostic va nécessiter une collaboration pluridisciplinaire où l’ophtalmologiste devra rechercher, prévenir et prendre en charge les complications oculaires.
Morphologie orbitofaciale (fig. 6-11) : la déformation induite du crâne (± de la face) est le plus souvent caractéristique pour chaque type de craniosténose. La palpation de la suture atteinte objective l’existence d’un bourrelet ou d’une crête synostosique et non d’une dépression. Ce n’est pas une pathologie des fontanelles. La malformation s’évalue grâce aux mesures suivantes : diamètre intercanthal interne et externe, distance interpupillaire, distance oropalpébrale, longueur palpébrale, position des points lacrymaux. Ces mesures permettent de caractériser un éventuel pseudo-hypertélorisme et peuvent guider et évaluer la chirurgie craniofaciale. La configuration des fentes palpébrales (obliquité) est importante à vérifier, elle peut révéler une torsion des orbites associée à un syndrome A ou V. La symétrie des orbites est à vérifier de même qu’un torticolis est à rechercher.
Lorsqu’une exophtalmie est présente, l’exophtalmomètre de Hertel est utilisé de façon reproductible.
Cette étude est systématiquement mesurée sous cycloplégiques à l’aide d’un réfractomètre. Elle permet de mettre en évidence les anomalies réfractives amblyogènes.
Cette étude permet :
de préciser l’état sensoriel : recherche d’une amblyopie, étude de la fixation, analyse de la vision binoculaire en position de torticolis et en position primaire, mesure de la vision stéréoscopique ;
de rechercher une déviation et la mesurer en position primaire et dans le regard en haut et en bas à la recherche d’un syndrome alphabétique A ou V ;
d’évaluer la motilité dans toutes les directions du regard.
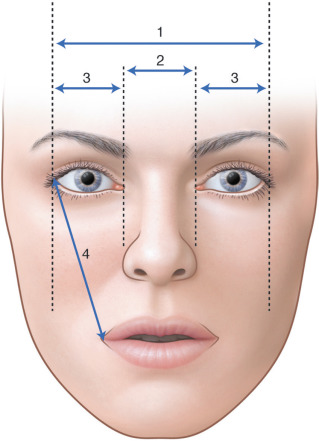
Fig. 6-11 Paramètres orbitofaciaux : points de mesure pour la distance intercanthale externe (1), la distance intercanthale interne (2), la longueur palpébrale (3) et la distance oropalpébrale (4).
L’examen à la lampe à fente analyse les segments antérieur et postérieur (afin de diagnostiquer une dysgénésie du segment antérieur associée) [8]. La mesure de la pression oculaire est effectuée. Le fond d’oeil (FO) vérifie l’état du nerf optique : lorsque la coopération est impossible (malgré anesthésie locale et écarteur palpébral), un examen sous anesthésie générale sera systématiquement programmé avec clichés du FO (RetCam™) permettant une iconographie et un suivi.
L’examen à la lampe à fente analyse les segments antérieur et postérieur (afin de diagnostiquer une dysgénésie du segment antérieur associée) [8]. La mesure de la pression oculaire est effectuée. Le fond d’oeil (FO) vérifie l’état du nerf optique : lorsque la coopération est impossible (malgré anesthésie locale et écarteur palpébral), un examen sous anesthésie générale sera systématiquement programmé avec clichés du FO (RetCam™) permettant une iconographie et un suivi.
Effectué en mode retinal nerve fiber layer (RNFL) à partir de l’âge de 3 ans, cet examen est devenu indispensable pour détecter l’oedème papillaire débutant de façon précoce ou objectiver l’atrophie optique [9]. En revanche, il n’est pas assez performant pour différencier les drusen de la papille de l’oedème [10].
C’est un examen anodin et facile à réaliser pour :
éliminer des drusen papillaires [11], notamment lorsqu’elles sont profondes et confondues avec un oedème papillaire. Certains auteurs ont démontré la supériorité de l’échographie en mode B par rapport aux clichés en autofluorescence de la papille, l’OCT n’étant pas encore performante pour trancher entre les deux diagnostics. Aujourd’hui, c’est l’examen de choix dans le diagnostic des drusen de la papille chez l’enfant ;
détecter des anomalies musculaires au cours des craniosténoses telles une épaisseur excessive, une finesse trop importante, une agénésie, une fibrose ainsi qu’un état cicatriciel postopératoire. L’intérêt principal de cette échographie est de mieux guider la chirurgie du strabisme dans les cas complexes [12].
L’iconographie joue un rôle important pour dresser un bilan lésionnel, suivre l’évolution des lésions, apprécier le résultat d’une chirurgie correctrice et a un intérêt médico-légal. Ses champs d’application sont :
le FO (atteinte du nerf optique, pseudo-ectopie maculaire) soit par rétinographie dès l’âge de 3 ans, soit par la RetCam™ avant cet âge ;
la photographie du regard (déviation oculaire avant et après chirurgie craniofaciale ou oculomotrice) ;
les déformations morphologiques du visage et du crâne avant et après chirurgie craniofaciale.
L’électrophysiologie sera programmée au cas par cas en fonction du doute sur l’atteinte associée du nerf optique.
C’est un examen invasif et de réalisation potentiellement difficile chez l’enfant mais indispensable pour diagnostiquer un oedème papillaire en montrant la diffusion du produit de contraste autour du nerf optique qu’il y ait ou non la présence de drusen associées.
Le bilan d’imagerie par tomodensitométrie cérébrale et craniofaciale est essentiel à la réalisation du plan neurochirurgical. L’imagerie par résonance magnétique (IRM) cérébrale intervient en seconde intention dans les formes complexes, syndromiques ou en cas d’HTIC.
Dans la plupart des cas, le diagnostic de craniosténose est un diagnostic clinique qui nécessite un bilan ophtalmologique précoce et un suivi rigoureux. Les examens complémentaires recherchent les complications, participent à la prise en charge thérapeutique et permettent de suivre l’évolution.
Les conséquences oculaires dépendent du caractère isolé ou syndromique de la craniosténose, de sa sévérité et de la précocité de la chirurgie craniofaciale.
Elles sont le plus souvent secondaires aux modifications des structures osseuses au niveau :
du crâne, la synostose entraînant une HTIC qui secondairement altère le nerf optique ;
de l’orbite avec répercussion directe sur le globe oculaire.
Elles sont plus rarement en rapport avec des malformations développementales à type d’anomalies des muscles oculomoteurs, d’anomalies rétiniennes comme dans le syndrome de Stickler et le syndrome de Duane associé à la malformation craniofaciale [1].
Ces complications, souvent associées entre elles, sont à type d’atteinte du nerf optique, de troubles réfractifs, de troubles oculomoteurs, d’exorbitisme (pseudo-exophtalmie) et souffrance cornéenne et de conjonctivite. Leur prise en charge doit être le plus précoce possible, en raison de leur menace potentielle visuelle.
L’oedème papillaire et l’atrophie optique post-stase peuvent être présents dès la naissance ou progresser secondairement, impliquant un dépistage systématique du FO et des examens complémentaires (OCT, échographie, potentiels évoqués visuels [PEV], angiographie rétinienne et rétinographie). L’atteinte papillaire augmente avec le nombre, la localisation des sutures concernées (cintre bicoronal) et le caractère syndromique de la craniosténose. Ainsi, dans les craniosténoses syndromiques [13], la fréquence est plus élevée que dans les craniosténoses non syndromiques [14].
L’atteinte du nerf optique est liée principalement à la présence d’une HTIC, plus rarement d’autres facteurs tels un canal optique anatomiquement anormal :
HTIC :
l’augmentation de la pression intracrânienne secondaire à la soudure des sutures s’oppose à la croissance de l’encéphale surtout lors de sa phase de croissance rapide, au cours des deux premières années de vie. Toutefois, une HTIC secondaire est à redouter tout au long de la croissance crânienne, même chez les patients opérés, ce qui implique une vigilance jusqu’à la fin de l’adolescence ;
l’oedème papillaire, qui peut être suivi d’une atrophie optique post-stase, est la manifestation la plus évidente de l’HTIC. Il correspond à une infiltration interstitielle des fibres nerveuses et peut progresser du simple flou des bords de la papille à la stase papillaire. L’oedème papillaire de stase peut se développer précocement et brutalement indiquant une intervention neurochirurgicale urgente (voir fig. 6-9g, h) pour éviter une perte de vision irréversible : il peut être le seul signe témoignant de l’HTIC [15]. La mesure de la pression intracrânienne jugée comme invasive n’est plus systématique ;
au stade d’atrophie optique, l’atteinte peut évoluer de la pâleur discrète, difficile à mettre en évidence, à l’atrophie optique totale à bord net avec cécité. Elle peut être uni- ou bilatérale, révéler une HTIC précoce passée inaperçue, une HTIC chronique probablement responsable du retard mental rencontré dans des formes sévères diagnostiquées tardivement.
anomalie du canal optique : la compression du nerf optique dans un canal optique étroit, déformé, coudé ou rétréci peut se révéler par une atrophie optique. Dans certains cas, une hypoplasie primitive du nerf optique peut être observée [2].
Sur le plan thérapeutique, la correction neurochirurgicale précoce, avec expansion volumique de la boîte crânienne, permet de prévenir ou traiter l’HTIC déjà présente.
Face à l’amblyopie organique par atteinte du nerf optique, une rééducation est à entreprendre pour éliminer la part fonctionnelle de l’amblyopie.
Des troubles réfractifs sont retrouvés très fréquemment. Dans une série de craniosténoses non syndromiques, Chung [16] les retrouve dans 51 % des cas (45/88) à type d’astigmatisme (35 % ), d’hypermétropie (27 % ), de myopie (5 % ), d’anisométropie (20 % ) avec une plus forte probabilité pour les synostoses coronales.
Dans les craniosténoses syndromiques, l’astigmatisme est bilatéral, plus symétrique et plus sévère pouvant aller jusqu’à 4-5 D [17]. L’astigmatisme peut avoir plusieurs origines :
origine orbitaire acquise due à la craniosténose, provoquée par les tractions anormales exercées sur la périorbite qui se répercutent sur la partie antérieure du globe oculaire ;
par diminution entravée de l’astigmatisme physiologique due à la déformation osseuse.
L’anisométropie est également fréquente.
Ces anomalies réfractives doivent être recherchées et corrigées le plus tôt possible pour ne pas rajouter une part fonctionnelle à l’amblyopie par atteinte du nerf optique. Elles sont le plus souvent bilatérales symétriques ou asymétriques et fonction de la craniosténose. Ces amétropies peuvent soit diminuer après correction chirurgicale précoce [14] soit perdurer [15]. Sur le plan de la correction optique, l’adaptation de la monture et le port peuvent être entravés par la malformation orbitofaciale.
La fréquence du strabisme est très variable dans la littérature, mais reste globalement très importante dans les craniosténoses isolées et syndromiques. Le strabisme retrouvé est plus souvent horizontal avec syndrome V, en dehors des plagiocéphalies antérieures où il est vertical. L’origine est orbitaire et/ou musculaire.
Dans les plagiocéphalies synostotiques antérieures, le strabisme vertical avec torticolis est présent dans 57,6 à 66 % [16–19]. La tomodensitométrie en 3 dimensions (TDM 3D) montre une déformation unilatérale de la région frontozygomatique avec récession de cette zone et position postérieure de l’apophyse frontale qui est à l’origine de tractions sur les muscles et fascias orbitaires [17]. Il en résulte le plus souvent un strabisme vertical (fig. 6-5e) simulant une paralysie de l’oblique supérieur du côté atteint avec torticolis controlatéral, associé également souvent à un élément horizontal (voir fig. 6-5h-k). Le strabisme peut aussi survenir du côté opposé à la suture synostosée attestant que le côté dit sain n’est en fait pas normal du fait des phénomènes de croissance compensatrice [19, 20]. Après chirurgie craniofaciale (avancement fronto-orbitaire), ce strabisme vertical et horizontal peut diminuer [16], voire disparaître [15] mais aussi survenir en postopératoire [17]. Il est donc primordial d’en informer les parents et de suivre ces enfants sur le plan oculomoteur.
Dans les craniosténoses syndromiques, le strabisme est également très fréquent [20, 21], horizontal (très souvent divergent : fig. 6-10) associé à un syndrome alphabétique en V [22] allant pour Rosenberg [23] de 39 à 90,9 % pour les syndromes de Crouzon, d’Apert, Pfeiffer et de Saethre-Chotzen.
Le strabisme lors du syndrome de Crouzon est le plus souvent divergent avec syndrome V (voir fig. 6-9a, b) [21] et lié à l’extorsion des orbites. L’extorsion orbitaire modifie l’action des muscles qui deviennent fonctionnellement des muscles obliques, le muscle droit latéral s’insérant plus bas que le droit médial. Au cours du syndrome d’Apert, l’hypertélorisme est important et l’extorsion est moindre, la déviation horizontale associée est plus volontiers convergente (voir fig. 6-12a avant et fig. 6-12b après chirurgie oculomotrice effective) mais toujours avec un syndrome V avec hyperaction de l’oblique inférieur.
Le syndrome alphabétique en V associé à l’élément horizontal est le plus fréquent avec exotropie dans le regard en haut, orthotropie ou ésotropie dans le regard en bas (fig. 6-13a-c à 4 ans et fig. 6-13e-g à l’âge adulte). L’élévation en adduction est variable allant de modérée à très sévère et la limitation de l’abaissement en adduction va de léger à modérée. Il n’existe pas de cause unique expliquant le syndrome V. Les différentes explications retrouvées dans la littérature font état : d’une dysfonction innervationnelle (théorie des muscles horizontaux, verticaux ou obliques), d’une étiologie anatomique (désagittalisation ou anomalie musculaire) ou plus simplement d’une torsion orbitaire [22].
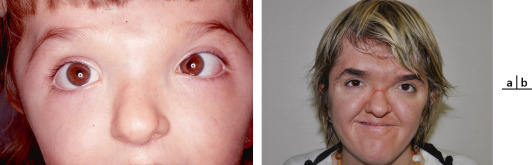
Fig. 6-12 Syndrome d’Apert.
a. Strabisme convergent chez un enfant de 4 ans porteur d’un syndrome d’Apert. b. Après chirurgie craniofaciale puis chirurgie du strabisme à l’âge adulte.
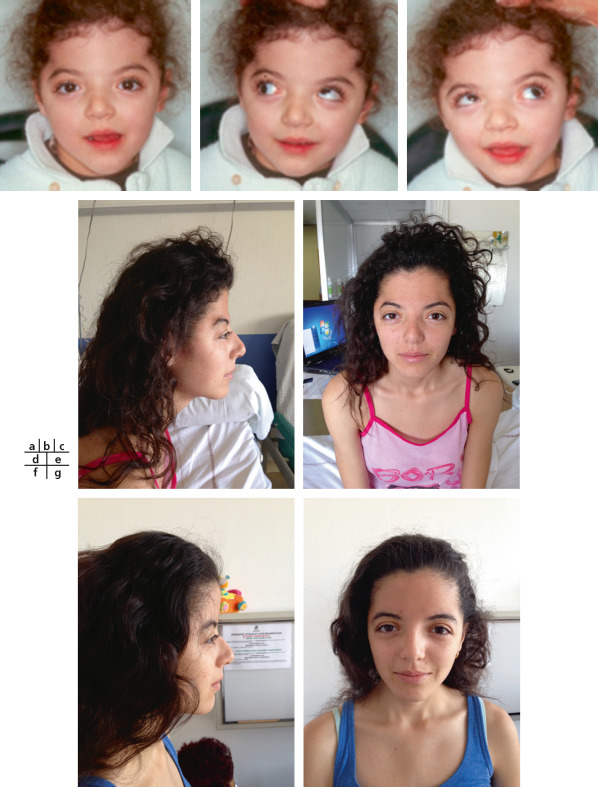
Fig. 6-13 Évolution d’un syndrome de Crouzon.
a-c. Enfant âgée de 4 ans, opérée à l’âge de 9 mois par avancée frontale ; présence d’un syndrome V avec orthotropie en position primaire. d, e. Cette même enfant, perdue de vue jusqu’à l’âge de 16 ans, reconsulte pour céphalées chroniques (persistance d’un morphotype de brachycéphalie). f, g. La réintervention craniofaciale d’avancée fronto-orbitaire a permis la disparition des céphalées et une amélioration esthétique.
Les anomalies retrouvées concernent le droit supérieur, l’oblique supérieur, l’oblique inférieur et le droit médial. Il s’agit d’anomalies d’insertion, de structure (micro-anatomie, muscle bifide, muscles fusionnés), d’anomalies d’orientation (localisation anormale dans position aberrante) et d’agénésie de certains muscles (droit supérieur, oblique supérieur) [12, 23]. Ces anomalies sont plus fréquentes dans les craniosténoses syndromiques.
La chirurgie du muscle oblique supérieur ou inférieur dans la pseudo-paralysie de l’oblique supérieur répond bien aux techniques classiques.
Pour le syndrome V, la chirurgie des obliques inférieurs hyperactifs apparaît être un traitement qui améliore mais ne normalise pas la motilité dans ce type de pathologies [24].
Quelle que soit la chirurgie indiquée chez ces patients, il est important de vérifier la présence et l’état des muscles oculomoteurs par échographie 3D et IRM pour aider au plan chirurgical [12].
L’amblyopie fonctionnelle strabique peut s’additionner à une amblyopie fonctionnelle réfractive par astigmatisme ou anisométropie. Son traitement répond bien aux méthodes classiques actuelles de rééducation. Ce traitement peut être effectué avant ou après chirurgie craniofaciale correctrice. Le but est l’obtention de la meilleure acuité visuelle permettant si possible le développement de la fusion binoculaire.
L’installation de la vision stéréoscopique (4-5e mois) et le développement de la vision monoculaire (10 ans) peuvent être entravés par un strabisme secondaire à la malformation orbitaire et empêcher le développement d’une vision binoculaire normale. Ainsi, la majorité de ces strabismes s’accompagnent d’une absence de correspondance rétinienne [25].
Complication des craniosténoses syndromiques, la pseudo-exophtalmie relève d’un rapport anormal entre la cavité orbitaire et son contenu par limitation de la profondeur des orbites pouvant se compliquer d’un véritable exorbitisme. Cet exorbitisme peut être très marqué dans le syndrome de Crouzon où il peut s’aggraver jusqu’à la luxation du globe oculaire spontanément ou par manipulation de la paupière, ou par collyre à base de néosynéphrine. Pour un même syndrome, le degré d’exorbitisme varie d’un patient à l’autre. Imai [26] en 2013, en comparant le volume orbital d’un groupe de syndrome de Crouzon et d’un groupe de syndrome d’Apert, montre que le volume orbital est le plus faible dans le syndrome de Crouzon ; l’exophtalmie et le strabisme divergent ne sont présents que dans le groupe de syndrome de Crouzon. Dans les formes sévères, associant atteintes cornéennes d’exposition et/ ou atteintes du nerf optique, une cranioplastie en urgence consistant en une expansion de l’orbite ne rétablit pas entièrement le volume orbital normal. Mais dans la plupart des cas, il est utile pour soulager les symptômes à type d’exophtalmie, d’érosion de la cornée, de conjonctivite permettant d’éviter le risque de cécité.
Les points clés à connaître dans le cadre d’une craniosténose sont développés dans l’encadré ci-dessous et les tableaux 6-3 et 6-4.
À retenir – Complications ophtalmologiques des craniosténoses
Atteinte du nerf optique
Atteinte réfractive
Atteinte oculomotrice
Exophtalmie (exposition cornéenne)
Tableau 6-3 – Diagnostic, conséquences, prise en charge.
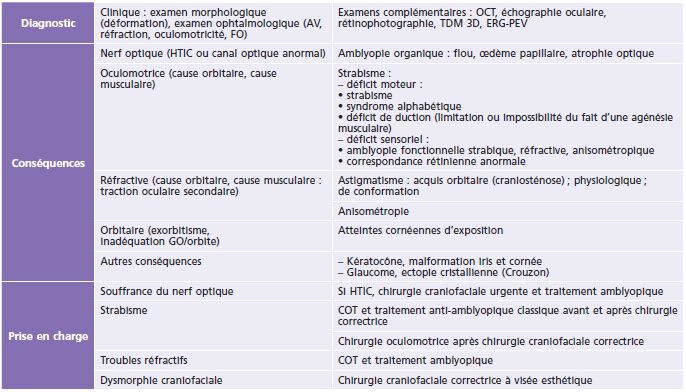
AV : acuité visuelle ; COT : correction optique totale ; ERG : électrorétinogramme ; FO : fond d’oeil ; GO : globe oculaire ; HTIC : hypertension intracrânienne ; OCT : optical coherence tomography ; PEV : potentiels évoqués visuels ; TDM 3D : tomodensitométrie en trois dimensions.

La chirurgie craniofaciale doit avoir trois objectifs dans la prise en charge des craniosténoses :
traiter le conflit entre l’encéphale et ses enveloppes ostéodurales, et prévenir l’installation d’une HTIC chronique ;
restaurer la symétrie orbitaire et traiter un hypertélorisme ou un hypotélorisme pour préserver un pronostic de vision binoculaire ;
améliorer le pronostic esthétique de ces malformations. Cet aspect ne doit pas être négligé étant donné l’impact sociétal de ces malformations du crâne et de la face.
Il faut distinguer l’attitude stéréotypée adoptée pour les craniosténoses simples dans la première année de vie avec une démarche et un calendrier chirurgical reproductible d’un patient à l’autre, et la prise en charge multidisciplinaire des craniosténoses complexes et craniofaciosténoses qui pourra comporter plusieurs gestes chirurgicaux à des âges variables en fonction de la clinique et de la sévérité de l’atteinte.
L’intervention est idéalement réalisée à 3 ou 4 mois de vie. Le principe est de pratiquer une craniectomie « en H » libérant les volets temporopariétaux. Ce geste peut être effectué par méthode conventionnelle au moyen d’une incision bicoronale postérieure ou par méthode endoscopique par deux incisions bregmatique et lambdatique. Le remodelage de la voûte est ici passif car obtenu secondairement par l’appui postérieur et la poussée encéphalique.
L’intervention est réalisée dans la première année de vie, généralement autour de 9 mois. Elle consiste en un remodelage actif et complet du front et du bandeau orbitaire avec un avancement latéral du bandeau afin d’ouvrir l’angle formé par les cadres orbitaires supérieurs au niveau du nasion.
La planification de l’opération se fait également dans la première année de vie (9 mois) avec un remodelage asymétrique du front et des cadres orbitaires supérieurs et un avancement unilatéral du bandeau orbitaire. Cette intervention ne modifie pas la scoliose faciale qui se corrige secondairement avec la croissance et la prise en charge ophtalmologique du strabisme.
La recherche de signes associés en faveur d’une étiologie syndromique doit prévaloir. Une surveillance ophtalmologique rapprochée à la recherche de signes d’HTIC doit être effectuée dans les mois qui précèdent la chirurgie d’avancée fronto-orbitaire. En l’absence d’oedème papillaire, on attend l’âge de 9 mois pour intervenir.
La stratégie à long terme consiste à accompagner la croissance encéphalique de l’enfant. Il est généralement nécessaire d’envisager une expansion volumique du crâne de l’enfant pour traiter et prévenir l’HTIC et ses conséquences (ophtalmologiques, malformation de Chiari). La tendance actuelle est de privilégier une expansion postérieure précoce par distraction ostéogénique de la voûte postérieure [27], et de pratiquer une avancée frontofaciale monobloc secondairement, également par distraction ostéogénique.
La déformation posturale postérieure appelée communément « plagiocéphalie postérieure » est responsable d’une déformation crânienne asymétrique très fréquente depuis la publication en 1992 de l’American Academy of Pediatrics recommandant de faire dormir le nourrisson sur le dos. Elle peut avoir un retentissement fronto-orbitaire caractéristique (fig. 6-14). C’est un diagnostic différentiel essentiel car cette affection positionnelle n’entre pas dans le cadre nosologique des craniosténoses. Son diagnostic doit rester clinique sans imagerie et la prise en charge non chirurgicale repose sur des mesures positionnelles. Les facteurs de risque sont : des contraintes in utero expliquant leur plus grande fréquence dans les grossesses multiples, une position de sommeil en décubitus dorsal, un torticolis et une prématurité.
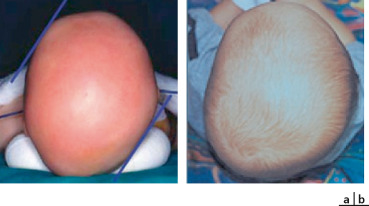
Fig. 6-14 Déformation positionnelle postérieure (a : aplatissement occipital), communément appelée « plagiocéphalie postérieure » , expliquant le retentissement antérieur frontoorbitaire (b : avancée de la bosse frontale homolatérale).
Les craniosténoses sont des affections rares, potentiellement cécitantes dont le diagnostic et la prise en charge doivent être précoces. La responsabilité directe de la malformation craniofaciale dans la survenue des complications oculaires et neurologiques souligne l’indispensable collaboration entre neurochirurgien et ophtalmologiste. Ces deux spécialistes, acteurs de la réhabilitation visuelle et esthétique de l’enfant, oeuvrent conjointement pour donner à ce dernier la place qui lui est due dans la société.
[1] Crompton J, Black J. Craniofacial abnormalities. In : Taylor D, Hoyt CS. Pediatric ophthalmology and strabismus. 4th ed. London : Elsevier ; 2013, chap. 28, p. 243-64.
[2] Miller MT, Shah HA, Nexlin A. Craniofacial syndromes and malformations. In : Wright KW, Spiegel PH. Pediatric ophthalmology and strabismus. 3rd ed. Springer ; 2015, p. 1095-38.
[3] Di Rocco F, Arnaud E, Renier D. Evolution in the frequency of nonsyndromic craniosynostosis. J Neurosurg Pediatr 2009 ; 4(1) : 21-5.
[4] Apert E. De l’acrocéphalosyndactylie. Soc Med, Paris 1906 ; 23 : 1310-30.
[5] Crouzon O. Dysostose craniofaciale héréditaire. Bull Mem Soc Med Hop Paris 1912 ; 33 : 545-55.
[6] Pfeiffer RA. Dominant hereditary acrocephalosyndactylia. Z Kinderheilkd 1964 ; 90 : 301-20.
[7] Sharma A, Patel N, Arora S, Ramachandran R. Child with Saethre-Chotzen syndrome : anesthetic management and literature review. Acta Anaesthesiol Belg 2014 ; 65 : 179-82.
[8] Okajima K, Robinson LK, Hart MA, et al. Ocular anterior chamber dysgenesis in craniosynostosis syndromes with a fibroblast growth factor receptor 2 mutation. American Journal of Medical Genetics 1999 ; 85 : 160-70.
[9] Dagi LR, Tiedemann LM, Heidary G, et al. Using spectral-domain optical coherence tomography to detect optic neuropathy in patients with craniosynostosis. J AAPOS 2014 ; 18 : 543-9.
[10] Kulkarni KM, Pasol J, Rosa PR, Lam BL. Differentiating mild papilledema and buried optic nerve head drusen using spectral domain optical coherence tomography. Ophthalmology 2014 ; 121 : 959-63.
[11] Naoum S, Bouacha I, Drumare I, et al. Druses de la papille de l’enfant : intérêt des différents examens d’imagerie. Journal Français d’Ophtalmologie 2016 ; 39 : 341-5.
[12] Somani S, MacKeen LD, Morad Y, et al. Assessment of extraocular muscles position and anatomy by 3-Dimensional ultrasonography : a trial in craniosynostosis patients. J AAPOS 2003 ; 7 : 54-9.
[13] Bannink N, Joosten KF, Van Veelen ML, et al. Papilledema in patients with Apert, Crouzon, and Pfeiffer syndrome: prevalence, efficacy of treatment and risk factors. J Surg Craniofac 2008 ; 19 : 121-7.
[14] Denis D, Genitori L, Bardot J, et al. Ocular findings in trigonocephaly. Graef Arch Clin Exp Ophthalmol 1994 ; 232 : 728-33.
[15] Bartels MC, Vaandrager JM, de Jong TH, Simonsz HJ. Visual loss in syndromic craniosynostosis with papilledema but without other symptoms of intracranial hypertension. J Craniofac Surg 2004 ; 15 : 1019-22.
[16] Chung SA, Yun IS, Moon JW, Lee JB. Ophthalmic findings in children with nonsyndromic craniosynostosis treated by expansion cranioplasty. J Craniofac Surg 2015 ; 26 : 79-83.
[17] Denis D, Genitori L, Conrath J, et al. Ocular findings in children operated on for plagiocephaly and trigonocephaly. Child’s Nerv Syst 1996 ; 12 : 683-9.
[18] Samra F, Paliga JT, Tahiri Y, et al. The prevalence of strabismus in unilateral coronal synostosis. Childs Nerv Syst 2015 ; 31 : 589-96.
[19] Macintosh C, Wall S, Leach C. Strabismus in unicoronal synostosis : ipsilateral or contralateral ? J Surg Craniofac Mai 2007 ; 18 : 465-9.
[20] Beckett JS, Persing JA, Steinbacher DM. Bilateral orbital dysmorphology in unicoronal synostosis. Plast Reconstr Surg 2013 ; 131 : 125-30.
[21] Denis D, Conrath J, Genitori L. Exophtalmie, astigmatisme et strabisme dans les cranio-faciosténoses : syndrome d’Apert et syndrome de Crouzon. Ophtalmologie 1997 ; 11 : 28-33.
[22] Ron Y, Dagi LR. The etiology of V-pattern strabismus in patients in craniosynostosis. Int Ophthalmol Clin 2008 ; 48 : 215-23.
[23] Rosenberg JB, Tepper OM, Medow NB. Strabismus in craniosynostosis. J Pediatr Ophthalmol Strabismus 2013 ; 50 : 140-8.
[24] Qiao T, Wang G, Xiong J, et al. Surgical treatment of V-pattern exotropia in Crouzon syndrome. J pediatric Ophthalmol Strabismus 2015 ; 52 : 299-304.
[25] Nelson LB, Ingoglia S, Breinin GM. Sensorimotor disturbances in craniostenosis. J Pediatr Ophthalmol Strabismus 1981 ; 18 : 32-41.
[26] Imai K, Fujimoto T, Takahashi M, et al. Preoperative and postoperative orbital volume in patients with Crouzon and Apert syndrome. J Surg Craniofac 2013 ; 24 : 191-4.
[27] Arnaud E, Marchac A, Jeblaoui Y, et al. Spring-assisted posterior skull expansion without osteotomies. Childs Nerv Syst 2012 ; 28 : 1545-9.
N. Dégardin, A. Gallucci, C. Jaloux, G. Pech-Gourg, D. Denis
Les malformations craniofaciales sont des malformations congénitales complexes et rares nécessitant une prise en charge pluridisciplinaire. La place de l’ophtalmologiste dans la prise en charge de ces malformations est importante en raison de la fréquence de l’atteinte orbitopalpébrale. Le spectre des fentes faciales nous intéresse plus particulièrement ici : ces fentes sont dites « rares » en opposition aux fentes labio-maxillo-palatines beaucoup plus fréquentes mais qui restent limitées au palais primaire et secondaire et qui n’ont donc pas de retentissement orbitaire et/ou palpébral.
Deux grands types de fentes faciales rares sont distingués :
les atteintes médianes qui influencent l’écart existant entre les orbites par excès – hypertélorisme – ou par défaut – hypotélorisme ;
les fentes latérofaciales ou faciales obliques passent dans l’aire orbitaire et entraînent de façon directe des anomalies des annexes de l’oeil ainsi qu’une malformation de la région orbitopalpébrale (interruption, distorsion, déplacement des structures orbitopalpébrales).
Les bourgeons faciaux constitutifs de la face embryonnaire ou bourgeons faciaux primordiaux, dérivant du mésenchyme de la crête neurale, apparaissent à la 4e semaine de développement et sont au nombre de cinq (fig. 6-15) :
le bourgeon (naso-)frontal, impair et médian, flanqué de deux renflements latéraux correspondant aux placodes olfactives ;
les deux bourgeons maxillaires (BMAX) qui s’immiscent entre le bourgeon frontal et les bourgeons mandibulaires délimitant latéralement le stomodæum ou bouche primitive ;
les deux bourgeons mandibulaires (BMAND).
De la 5e à la 7e semaine, la face va se constituer par développement et confluence des différents bourgeons.
À la 5e semaine, le bourgeon frontal (BF) ou nasofrontal se différencie en bourgeon nasal externe (BNE) et bourgeon nasal interne (BNI) autour de chaque placode olfactive.
La fusion des bourgeons (6e et 7e semaines) suit un ordre particulier :
fusion entre les deux bourgeons BMAND créant le menton et la lèvre inférieure ;
les deux BNI se rapprochent (poussés par la pression latérale des BMAX) et fusionnent entre eux sur la ligne médiane créant ainsi le philtrum de la lèvre supérieure ;
BNI et BMAX fusionnent pour former la lèvre supérieure (ligne de fusion = crête philtrale). Le BNE forme l’aile narinaire. La fusion entre BNI, BNE et BMAX donne naissance à l’aile narinaire ;
la fusion entre BNE et BMAX participe à la formation du massif facial latéral. Le canal lacrymonasal persiste au niveau de leur jonction ;
fusion entre BMAX et BMAND créant les commissures labialeset une partie des joues.
Donc, le front, le nez et le tiers médian de la lèvre supérieure dérivent du bourgeon frontonasal, les secteurs latéraux dépendent essentiellement des bourgeons maxillaire et mandibulaire (Tableau 6-5).
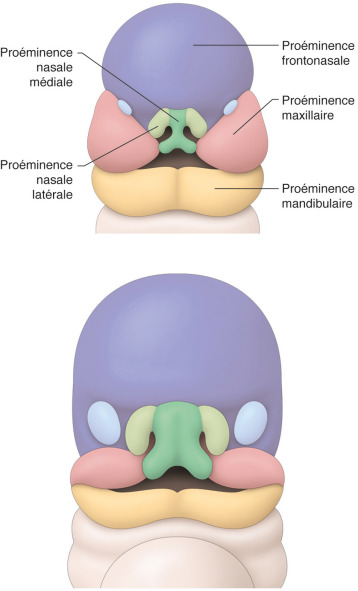
Fig. 6-15 Embryologie des bourgeons de la face.
Les malformations peuvent affecter uniquement le secteur frontonasal ou de façon associée les secteurs latéraux. Un défaut de fusion sur la zone de jonction entre deux secteurs entraîne une fente dont le trajet correspond aux zones de fusion décrites ci-dessus. Ainsi, un défaut de fusion entre les deux BNI sera responsable d’une fente labiale médiane, un défaut de fusion entre BNI et BMAX d’une fente labiale paramédiane sur la crête philtrale ou en dehors de celle-ci, un défaut de fusion BMAX et BMAND sera responsable d’une fente commissurale ou macrostomie.
D’autres fentes peuvent apparaître en dehors des zones de fusion. Leur étiopathogénie n’est pas clairement expliquée. Certains évoquent une origine amniotique, d’autres un processus de dysplasie foetale primitive d’origine vasculaire ou par défaut de migration des crêtes neurales [1].
La classification proposée par Paul Tessier est la plus communément admise (fig. 6-16) [2]. Il s’agit d’un système de classification anatomique dans lequel un numéro est attribué à chaque fente sur la base de sa position par rapport à la ligne médiane sagittale et à l’orbite. Elle est proposée selon une numérotation de 0 à 14 selon la position de la fente. Chaque axe a une composante crânienne (numéros 9 à 14) et une composante faciale (numéros 0 à 8). La fente atteint les parties molles et l’os sous-jacent (fig. 6-17 : fente 3).
Les fentes médianes sont les fentes 14-0, 13-1, 12-2. Ces fentes entraînent un hypertélorisme et nécessitent une réparation nasale.
Les fentes latérales sont les fentes 11-3, 10-4, 9-5, 6, 7, 8. Ces fentes entraînent un colobome palpébral et nécessitent une réparation des paupières et du défect orbitaire.
Tableau 6-5 – Ébauches contributives dans la formation de la face.
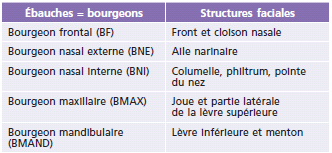
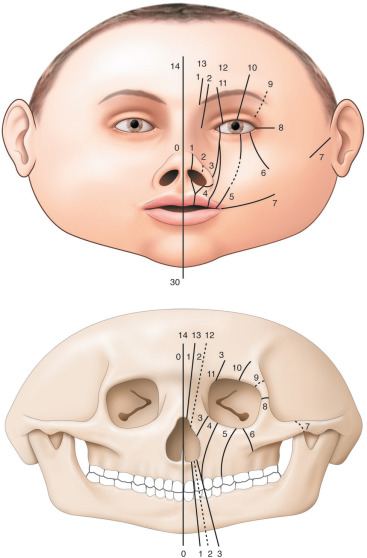
Fig. 6-16 Schématisation des fentes faciales rares selon la classification de Tessier.
(Source : O. Galatoire, Chirurgie du regard, SFO, 2016.)
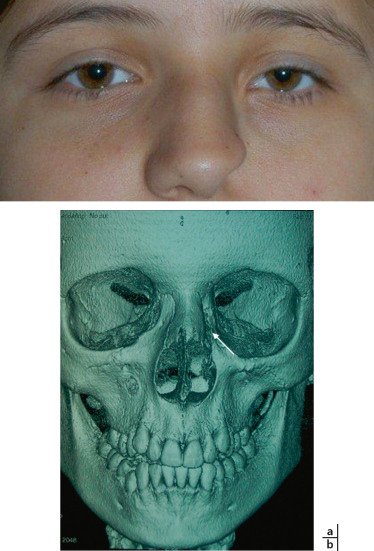
Fig. 6-17 Jeune adolescente présentant une fente faciale 3 selon la classification de Tessier non traitée à l’âge de 12 ans.
a. À l’examen clinique, la fente latéronasale déplaçant le pied d’aile narinaire s’associe à une dystopie canthale interne. b. L’imagerie (TDM du massif facial avec reconstruction tridimensionnelle) montre la fente latéronasale (flèche) au niveau de la branche montante du maxillaire.
Seules les fentes entraînant une répercussion oculo-orbitaire seront abordées :
fentes 0 et 14 : hypertélorisme ;
fente 3 (orbitomaxillaire interne) : microphtalmie, anomalies des voies lacrymales ;
fente 4 (orbitomaxillaire médiane) : malformation du plancher de l’orbite et du rebord orbitaire ;
fente 5 (orbitomaxillaire externe) : microphtalmie et anomalies du tiers moyen de la paupière inférieure ;
fente 6 (inter-zygomato-malaire) : colobome palpébral inférieur à la jonction tiers médian-tiers externe ;
fente 7 (temporozygomatique) : une obliquité en bas et en dehors de la fente palpébrale, hypoblépharie inférieure ;
fente 8 (frontozygomatique) : colobomes palpébraux, dystopie canthale latérale ;
fente 9 (orbitaire supéro-externe) : microphtalmie, anomalies de l’angle supéro-externe de l’orbite ;
fente 10 : colobome paupière supérieure et de l’iris, ablépharie.
D’autres classifications ont été proposées, fondées sur des considérations embryologiques [3, 4]. Pour Mazzola, le terme de fente devrait être réservé uniquement à la non-fusion des bourgeons faciaux réalisant les « vraies fentes » , comme l’absence de fusion entre le BN et le BMAX ou fente oro-naso-oculaire (= fente 3 de Tessier), ou entre les deux BNI (fente labiale médiane) ou encore entre le BMAND et BMAX : macrostomie. Pour toutes les autres anomalies en dehors des lignes de fusion embryonnaire, une altération des centres d’ossification embryonnaire serait primitivement à l’origine de la fente et il conviendrait alors de plutôt utiliser le terme de dysplasie : ainsi on devrait parler de dysplasie internasale, de dysplasie nasomaxillaire ou maxillaire médiale (fente 4), de dysplasie maxillaire latérale ou maxillaire et malaire (fente 5), selon le secteur primitivement concerné.
La classification de Fries [5] propose une classification régionale fondée sur le secteur d’atteinte orbitaire regroupant les « fentes » en fonction du secteur d’atteinte et donc des principales caractéristiques cliniques susceptibles d’être retrouvées :
secteur 1 : les fentes médianes internasales ou intranasales (équivalent fentes 0-14, 1-13 de Tessier) qui se traduisent par un hypertélorisme ;
secteur 2 : secteur nasolacrymal et canthal interne avec possibilité d’atteinte lacrymale (équivalent fentes 2-12, 3-11 de Tessier) ;
secteur 3 : ou secteur palpébral (équivalent fentes 4 et 5 de Tessier) avec présence de colobomes des paupières ;
secteur 4 : ou canthal latéral avec des dystopies canthales latérales (fentes 6, 7, 8 de Tessier).
Ces malformations concernent exclusivement ou majoritairement le secteur frontonasal et impliquent souvent le développement cérébral. Il en résulte sur les orbites un écart trop important de ceux-ci, l’hypertélorisme, ou au contraire un écart insuffisant de ceux-ci, l’hypotélorisme.
Les processus frontonasaux produisent non seulement la partie médiane de la face mais contribuent au développement du tube neural en hémisphère et bulbes optiques et olfactifs. Ainsi, l’holoprosencéphalie entraîne un défect cérébral mais également médiofacial avec l’existence d’un hypotélorisme. Les patients avec hypotélorisme ont plus de risques d’anomalies cérébrales associées que les hypertélorismes.
L’hypertélorisme est défini par un écart trop important entre les orbites osseuses : chez l’adulte par une distance interorbitaire (DIO) de plus de 34 mm. Cette distance limite est de 20 mm chez les nourrissons et 24 mm chez les enfants. Il est à différencier du télécanthus (déplacement latéral des canthi internes entraînant une fausse impression d’hypertélorisme). Bien souvent les deux sont en relation mais pas obligatoirement.
Les principales causes d’hypertélorisme congénital sont les craniosténoses concernant la voûte antérieure et/ou la base du crâne, les syndromes avec faciocraniosténoses (syndromes de Crouzon, d’Apert, de Pfeiffer, de Saethre-Chotzen : voir plus haut Craniosténoses), les méningo-encéphalocèles antérieures frontonasales ou fronto-ethmoïdales, les fentes faciales 0 à 4 et 11 à 14 de la classification de Tessier (fig. 6-18).
L’atteinte clinique dans ce groupe de fentes médianes est variable : de la simple bifidité de la pointe du nez à des formes beaucoup plus sévères avec duplication nasale et méningocèle fronto-orbitaire. Le signe constant est l’hypertélorisme plus ou moins important. L’hypertélorisme dans le cadre des fentes faciales est classé selon Kawamoto en cinq types du plus médian vers le plus latéral [6] :
groupe 1 : hypertélorisme lié à une craniosténose ;
groupe 2 : fente médiane ou dysraphie craniofaciale médiane (anomalie anatomique due à un défaut de fusion de deux structures) – fente 0-14 de Tessier – caractérisée par la symptomatologie clinique suivante : hypertélorisme, racine du nez élargie, crâne antérieur bifide, fente faciale médiane incluant le nez et/ou la lèvre et parfois le palais. Elle concerne l’os frontal et peut comporter une myéloméningocèle frontale médiane, l’ethmoïde (duplication de l’apophyse crista galli), le nez avec une duplication septale ou septocolumellaire, la lèvre supérieure (fente labiale médiane) et parfois un widow’s peak (implantation anormale de la ligne antérieure du cuir chevelu) (fig. 6-19) ;
groupe 3 : encéphalocèle frontale (hernie des lobes frontaux au niveau de la fente médiane au niveau de l’aire du sinus frontal de l’os frontal). Celle-ci épargne le tiers inférieur de la face mais impacte le tiers médian et supérieur : hypertélorisme, front large avec encéphalocèle et sourcils particulièrement écartés ;
groupe 4 : hypertélorisme lié à une fente paramédiane unilatérale (habituellement de type 1-13). Cet hypertélorisme est alors asymétrique ;
groupe 5 : fentes naso-oculaires (fentes 2, 3, 11 et 12 de Tessier). La position plus latérale de ces fentes provoque moins d’hypertélorisme mais davantage de dystopie canthale interne.
Il est intéressant de noter ici que les fentes médianes et paramédianes peuvent exister isolément ou s’associer à d’autres fentes faciales ou crâniennes plus latérales, responsables d’atteintes surajoutées à type de colobome des paupières. Ainsi, certaines associations sont rencontrées plus volontiers comme : 0-14, 4-10 et 5-9.
Les hypertélorismes moyens à importants font l’objet d’une prise en charge chirurgicale cranio-maxillo-faciale (fig. 6-20) [7, 8].
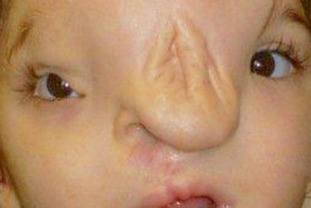
Fig. 6-18 Enfant âgé de 3 ans présentant une malformation craniofaciale complexe associant une fente médiane et paramédiane asymétrique avec aspect de fente 2 droite (fente labiale et nasoschisis) associée à un colobome palpébral supérieur médian droit et une dysplasie cranio-fronto-ethmoïdale.
Noter l’importance de l’hypertélorisme.
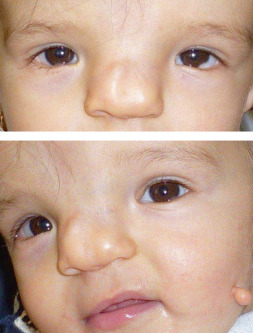
Fig. 6-19 Enfant âgée de 18 mois présentant un aspect de fente faciale médiane responsable d’un hypertélorisme, d’une bifidité de la pyramide nasale associée à plusieurs fistules nasales et paranasales dont une basicolumellaire médiane.

Fig. 6-20 Hypertélorisme associé à une anomalie d’Axenfeld chez un jeune garçon âgé de 2 ans.
L’hypotélorisme est défini par un écart insuffisant entre les orbites et est souvent associé à des malformations cérébrales (holoprosencéphalie pour exemple) (fig. 6-21). Il est rarement corrigé chirurgicalement.
Ces malformations impliquent, à la différence des fentes faciales médianes, de nombreuses structures osseuses (maxillaires, zygomatiques, mandibulaires), cutanées (palpébrales, jugales). Seules les fentes 3, 4 et 5, 6, 7, 8 selon la classification de Tessier seront étudiées.
La fente faciale 3 est également appelée dysplasie nasomaxillaire ou fente oro-naso-oculaire ou fente du secteur nasolacrymal et canthal interne [5].
Ces fentes sont un peu plus fréquemment rencontrées que les autres types de fentes faciales. Tous les cas connus sont sporadiques sans association syndromique ou prédilection de sexe. Cette fente implique la partie latérale du nez, le canthus interne et le système lacrymal sus-jacent. Elle peut être complète ou incomplète. Elle suit toujours le même trajet : du philtrum (un peu comme une fente labiale habituelle de la lèvre supérieure), au pourtour de l’aile narinaire (réalisant une distorsion ou un déplacement du pied de l’aile narinaire), puis atteint le canthus interne qui est toujours attiré vers le bas, raccourcissant la distance canthus- aile narinaire (fig. 6-22a). Si elle est associée à la fente homologue sur le versant crânien, soit la fente 11, elle touche le secteur médial de la paupière supérieure avec un colobome, la partie médiale du sourcil étant déplacée vers le bas, le front pouvant être également déformé. Il peut, par ailleurs, exister une fistule du canal lacrymal un peu plus bas et un peu plus latérale que le canthus dystopique (fig. 6-22b).
Au niveau osseux, il existe une atteinte de la gouttière lacrymale et du processus frontal du maxillaire (fig. 6-17).
Prise en charge : lorsqu’il existe une fente labiale ou une fente totale (éventualité très rare), la prise en charge est assez précoce, le plus souvent en condition néonatale. La reconstruction du canalv lacrymonasal est rarement possible. La dystopie canthale peut être corrigée dans le même temps ou ultérieurement.
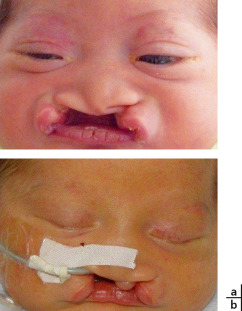
Fig. 6-21 Deux cas d’hypotélorisme avec agénésie prémaxillaire, holoprosencéphalie et fente labiale – unilatérale (a) et bilatérale (b) – chez deux nouveau-nés.
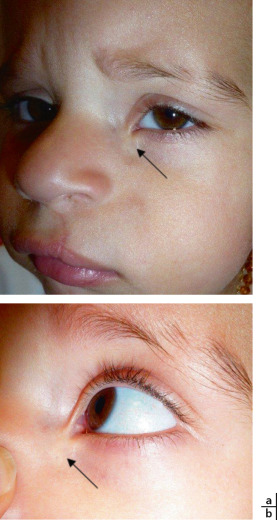
Fig. 6-22 Enfant de 2 ans présentant une fente faciale 3 de la classification de Tessier.
La fente labiale a été réparée en condition néonatale. Il persiste une dystopie canthale avec abouchement ectopique d’un canal lacrymal accessoire (flèche).
Les fentes faciales 4 et 5 sont également appelées fentes faciales latérales obliques ou fentes du secteur lacrymal (et leurs homologues sur le versant crânien : fentes 10 et 11) ou encore dysplasie maxillaire (médiale/latérale).
Ces fentes sont responsables d’une atteinte palpébrale à type de colobome plus ou moins associée à d’autres anomalies oculaires tels les symblépharons (fig. 6-23) [5, 9, 10].
La fente 4 épargne le plus souvent l’appareil lacrymal. Elle démarre au niveau de la lèvre supérieure latérale en dehors des crêtes philtrales, se poursuit latéralement par rapport aux ailes narinaires qui ne sont pas déformées et se termine à la jonction tiers interne et médian de la paupière inférieure.
La fente 5 part d’un point situé en dedans de la commissure labiale, traverse obliquement la joue et rejoint la région orbitopalpébrale à la jonction entre tiers médian et tiers latéral de la paupière inférieure. La paupière inférieure est souvent attirée vers le bas comme absorbée par la fente de la joue. Le contenu orbitaire descend dans la fente au travers de la fente osseuse maxillaire sous-jacente. Le globe oculaire peut être porteur d’anomalie à type de microphtalmie ou anophtalmie. Lorsque le globe est macroscopiquement normal, il existe néanmoins un risque majeur de kératopathie d’exposition pouvant compromettre la fonction visuelle en l’absence de protection efficace et de traitement chirurgical rapide [10, 11].
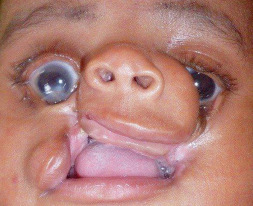
Fig. 6-23 Nourrisson âgé de 7 mois présentant des fentes faciales rares : de type 4 à droite et de type 5 à gauche.
Notons le symblépharon à droite avec une fonction visuelle déjà compromise de ce côté.
Les fentes latérofaciales 6, 7, 8 sont également appelées dysplasies otomandibulaires et syndromes des premier et second arcs branchiaux.
Les fentes 6, 7 et 8 de la classification de Tessier sont retrouvées de façon plus ou moins marquées dans l’ensemble des syndromes regroupés sous le terme de syndromes des premier et second arcs branchiaux.
La fente 6 chemine entre le zygoma et le maxillaire, elle associe :
un colobome palpébral inférieur à la jonction tiers médiantiers externe ;
un sillon sclérodermique vertical de la joue.
La fente 7 chemine entre l’os temporal et le zygoma, elle est donc responsable d’une atrophie :
de l’arcade zygomatique et du corps du zygoma ;
de la branche montante de la mandibule ;
d’une obliquité en bas et en dehors de la fente palpébrale.
On retrouve associés à ces malformations osseuses :
une microtie ;
des chondromes prétragiens ;
une macrostomie ;
une atrophie du muscle temporal ;
une anomalie de la patte des cheveux.
La fente 8 chemine entre l’os frontal et le zygoma, elle est donc responsable d’une atrophie :
de l’apophyse frontale du zygoma ;
de l’arcade zygomatique ;
de la branche montante mandibulaire.
On regroupe sous le nom de dysplasies otomandibulaires un certain nombre d’atteintes craniofaciales uni- ou bilatérales, congénitales, associant, à des degrés variables, malformation de l’oreille externe et parfois moyenne, hypoplasie de la branche montante mandibulaire, atteinte de l’os zygomatique et de l’arcade zygomatique. Nous aborderons les plus fréquentes : le syndrome de Goldenhar, le syndrome de Treacher-Collins et leurs répercussions ophtalmologiques.
Le syndrome de Goldenhar est le plus fréquent avec une incidence de 1/3 000 à 1/5 000 naissances par an. On parle parfois aussi de microsomie hémifaciale ou encore de spectre oculo-auriculo-vertébral. Il associe une atteinte craniofaciale le plus souvent unilatérale, parfois bilatérale asymétrique, à des atteintes vertébrales et parfois des malformations cardiaques et rénales.
Sur le plan facial, on retrouve le plus souvent [12, 13] :
une atteinte de l’oreille : externe, pouvant aller de la simple hypoplasie à une agénésie complète avec absence de conduit auditif externe et présence de fibrochondromes prétragiens ; moyenne et interne, entraînant une surdité de transmission et parfois même de perception ;
une atteinte de la branche montante mandibulaire responsable d’une asymétrie faciale avec déviation du menton du côté atteint et bascule du plan d’occlusion dentaire. La classification de Pruzanski [14] rend compte des différents types d’atteintes allant de la simple hypoplasie avec faible retentissement morphologique et fonctionnel, à une atteinte sévère concernant à la fois la région de l’angle et de la branche montante mandibulaire et toute l’articulation temporomandibulaire. Les formes sévères peuvent s’accompagner de détresse respiratoire néonatale pouvant aller jusqu’au recours à une trachéotomie ;
une macrostomie ;
une atteinte des muscles masticateurs et de la mimique, une agénésie parotidienne et une atteinte des structures nerveuses sensitives (troisième branche du trijumeau) et motrices (nerf facial), majorant d’autant plus l’asymétrie faciale ;
une fente palatine inconstante.
Sur le plan oculaire, on retrouve des dermoïdes épibulbaires constants, des colobomes palpébraux de localisation variable (fig. 6-24).
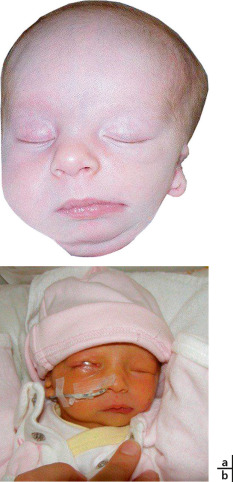
Fig. 6-24 Deux enfants présentant un syndrome de Goldenhar.
a. Nourrisson âgé de 1 mois avec atteinte hémifaciale gauche : macrostomie, anomalies du pavillon de l’oreille, déviation du menton vers la gauche (hypoplasie mandibulaire). b. Nouveau-né présentant un syndrome de Goldenhar avec hypoplasie mandibulaire gauche et colobome palpébral supérieur droit associés.
Le syndrome de Treacher-Collins, encore appelé syndrome de Franceschetti ou dysostose mandibulofaciale, est une atteinte génétique de transmission majoritairement autosomique dominante concernant une naissance sur 50 000. L’atteinte est essentiellement faciale, bilatérale, parfois asymétrique [12].
Dans le cas des syndromes de Treacher-Collins, l’atteinte est prédominante au niveau du tiers moyen de la face, l’hypoplasie zygomatique étant la plus marquée (fig. 6-25).
On retrouve ainsi :
un aspect rétrus de la pommette et des rebords infra-orbitaires ;
une obliquité en bas et en dehors des fentes palpébrales ;
une encoche jugale (sillon sclérodermique) ;
une atteinte mandibulaire moins marquée que dans le cas du Goldenhar (aspect effacé des angles mandibulaires, une brièveté des branches montantes responsable d’un profil dit concave) ;
une malformation de l’oreille externe ou moyenne ainsi que la présence de fibrochondromes ou de fistules pré-hélicéennes.
L’atteinte ophtalmologique est fréquente, elle peut concerner la taille du globe oculaire, la sclère et la conjonctive, les paupières, les voies lacrymales :
atteinte développementale du globe oculaire : microphtalmie et anophtalmie peuvent être retrouvées dans les syndromes des premier et second arcs branchiaux, même si elles sont relativement rares ;
atteinte scléro-conjonctivale :
dermoïdes épibulbaires [15, 16] : ils font partie de la triade initiale décrite par Goldenhar (avec les chondromes et fistules prétragiens). Il s’agit de tumeurs blanchâtres siégeant préférentiellement dans le quadrant inférotemporal du limbe. Il existe classiquement une lésion par oeil. Ces lésions peuvent être responsables d’un astigmatisme entraînant secondairement une amblyopie réfractive. Ces lésions peuvent augmenter de volume au cours de l’évolution : en cas de lésion étendue avec atteinte fonctionnelle visuelle, leur exérèse chirurgicale est indiquée ;
dermolipomes : ils sont retrouvés également dans les syndromes de Goldenhar, le plus souvent vers le canthus externe. En fonction de leur étendue, une imagerie peut être nécessaire pour préciser l’extension orbitaire, par échographie ou IRM.
atteinte palpébrale [15] : les colobomes sont fréquents, le plus souvent supérieurs dans le syndrome de Goldenhar, inférieurs dans le syndrome de Treacher-Collins. Il s’agit d’une agénésie du bord libre de la paupière avec absence de peau, de l’orbiculaire, du tarse, de la conjonctive, des cils et des structures glandulaires. Le pronostic est esthétique mais également fonctionnel en cas de colobome étendu, du fait du risque d’exposition cornéenne et d’ulcération de cornée ;
atteinte des voies lacrymales : épiphora par agénésie des voies lacrymales ;
atteinte des muscles oculomoteurs : le strabisme est fréquent. On observe une association fréquente au syndrome de Duane (avec agénésie du noyau de la Vie paire crânienne).
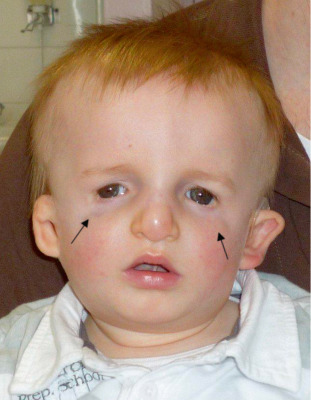
Fig. 6-25 Enfant âgé de 18 mois présentant un syndrome de Treacher-Collins.
Atteinte faciale bilatérale, anomalies des deux pavillons auriculaires, encoche malaire (flèches), obliquité en bas et en dehors des fentes palpébrales.
Il est important de demander un bilan ophtalmologique complet qui doit comporter un examen de la face, un examen des paupières/ voies lacrymales et du système oculomoteur (globe oculaire et muscles oculomoteurs).
Les paupières et les régions canthales sont assez couramment impliquées dans de nombreux syndromes craniofaciaux et notamment en cas de fente faciale. Les paupières doivent être soigneusement observées pour rechercher un épicanthus, un télécanthus, un ptosis, un entropion, un ectropion, un ou des colobomes ou un districhiasis.
Télécanthus et hypertélorisme doivent être notés et mesurés. Le télécanthus est mesuré par la distance intercanthale interne. La mesure de l’hypertélorisme est techniquement évaluée par la distance entre les parois osseuses internes orbitaires et nécessite donc la réalisation d’une imagerie osseuse (TDM). Une bonne estimation de la mesure de l’hypertélorisme est donnée par le calcul du ratio entre distance intercanthale et distance interpupillaire. Le rapport normal est d’environ 50 % .
Le déplacement vers le haut ou vers le bas de la position du tendon canthal interne définit la dystopie canthale interne. Le canthus latéral doit également être inspecté : des déviations verticales peuvent être notées. Normalement, le canthus latéral est positionné de 1 à 2 mm au-dessus du niveau horizontal du canthus interne. On parle d’orientation mongoloïde (verticale supérieure) ou antimongoloïde (verticale inférieure) des paupières.
L’évaluation exige un examen plus précis avec mesure de la hauteur de la fente palpébrale, position du pli palpébral supérieur et estimation de la fonction (course) du releveur de la paupière qui peut être difficile à préciser exactement chez le nourrisson. Chez les tout petits, les mesures ne peuvent être que très approximatives mais l’examen devient plus pertinent avec la coopération de l’enfant grandissant en fonction de l’âge.
On recherchera :
des colobomes palpébraux (largeur, exposition cornéenne)
un ptosis.
Le système lacrymal comporte des relations complexes entre l’os et les parties molles qui peuvent facilement être interrompues par une fente de type 3, par exemple, ou par une atteinte craniofaciale.
L’examen doit commencer par une inspection attentive de la position et de la perméabilité du point lacrymal et doit rechercher une anomalie osseuse sous-jacente. Le larmoiement ou épiphora est fréquent lors d’une anomalie craniofaciale (par atteinte ou anomalie lacrymale), soit dans le cadre du syndrome initial (par exemple fente craniofaciale 3), soit après un geste chirurgical craniofacial (par exemple après rapprochement orbitaire et canthopéxie transnasale dans une cure chirurgicale d’hypertélorisme).
Ce sont les atteintes craniofaciales qui impliquent la portion médiane de la face et la partie interne des paupières qui sont à risque de conséquences sur l’appareil lacrymonasal.
Un bilan ophtalmologique complet comprendra inspection, acuité visuelle, réfraction, lampe à fente, tonométrie-pachymétrie, FO et oculomotricité afin de diagnostiquer au plus tôt des troubles de la réfraction, des atteintes de la cornée, du nerf optique et des troubles oculomoteurs responsables d’amblyopie fonctionnelle et organique.
Les atteintes sont essentiellement oculaire (kératopathie d’exposition), palpébrale (ptosis), lacrymale et oculomotrice (strabisme, amblyopie réfractive et organique).
Le système visuel est menacé dans deux situations :
un colobome palpébral qui ne parvient pas à couvrir la cornée (cas fréquent dans les colobomes liés aux fentes 4 ou 5 de la classification de Tessier) ;
une exophtalmie sévère empêchant la fermeture des paupières (voir plus haut « Craniosténoses » ).
Le proptosis accompagne souvent les faciocraniosténoses tels les syndromes de Crouzon et d’Apert. La mesure précise avant la chirurgie avec l’exophtalmomètre de Hertel est indispensable pour évaluer l’effet de la chirurgie d’avancement orbitaire de même que l’examen de la papille à la recherche d’une souffrance.
De nombreux syndromes craniofaciaux s’accompagnent de perturbations de la vision binoculaire par strabisme qui doit être évalué le plus tôt possible.
La recherche de l’amblyopie doit être systématique chez tout patient porteur d’une anomalie craniofaciale. Le premier traitement proposé est alors la correction optique totale et l’occlusion monoculaire. L’adaptation de la monture de lunettes est souvent difficile du fait de la malformation craniofaciale. L’opticien, spécialisé doit fabriquer sur mesure la monture adaptée à chaque cas (fig. 6-26). Toutes les causes d’amblyopie doivent être recherchées : anisométropie, cataracte, taies cornéennes, ptosis, strabisme et atteinte du nerf optique. Le strabisme est très fréquent dans les syndromes craniofaciaux. Son traitement chirurgical doit être discuté. Il est souvent proposé après la chirurgie craniofaciale.
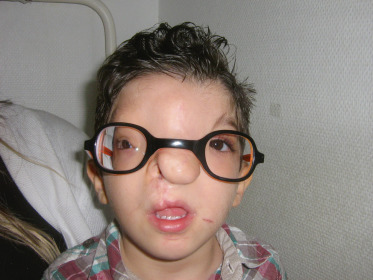
Fig. 6-26 Monture fabriquée sur mesure adaptée à la malformation craniofaciale.
La chirurgie est indiquée plus précocement lorsque le globe ou la vision sont menacés : oedème/atrophie optique, entropion, exposition cornéenne sévère ou ptosis sévère peuvent être des urgences chirurgicales. Le choix du type de réparation chirurgicale suit généralement les mêmes principes de base que chez les patients non porteurs de malformations craniofaciales congénitales.
Les indications essentielles sont l’exposition cornéenne (kératopathie d’exposition), les déformations importantes des canthi, les anomalies lacrymonasales, le ptosis, l’amblyopie. Toutes ces anomalies, lorsqu’elles s’expriment de façon sévère, peuvent conduire à la perte de la fonction visuelle.
La kératopathie d’exposition nécessite une prise en charge ophtalmologique immédiate avec collyres lubrifiants associés ou non à une tarsorraphie en attente de la chirurgie craniofaciale. Lorsque les colobomes sont limités et que la cornée n’est pas menacée, la chirurgie est reportée après l’âge de 6 mois. Les techniques de fermeture des colobomes comportent essentiellement des gestes de plasties locales de transposition ou de rotation ou encore des lambeaux hétéropalpébraux (contestés par certains auteurs car plus pourvoyeurs d’amblyopie). Lorsque la fonction protectrice palpébrale du globe est rétablie, des retouches pourront être envisagées dans un deuxième temps (épaisseur palpébrale et ptosis résiduel).
Deux grands types de techniques chirurgicales sont utilisés dans ces fentes médianes [7, 8, 12, 17] :
bipartition faciale : lorsqu’il existe une fente médiane ou paramédiane avec une obliquité latérale de l’axe des orbites. Elle est réalisée par voie d’abord coronale associée à une voie d’abord palpébrale ou conjonctivale et parfois une voie d’abord endobuccale permettant la réalisation des ostéotomies périorbitaires et la disjonction ptérygomaxillaire. Il n’y a pas d’ostéotomie entre maxillaire et orbite, les deux hémifaces étant mobilisées chacune en monobloc avec un mouvement de rotation en dedans, permettant de redresser l’axe des orbites en même temps que de rapprocher celles-ci (fig. 6-27) ;
– technique des « box » : mobilisation d’un ou des deux cadres orbitaires vers le dedans après résection du bloc ostéofibreux central (fig. 6-28).
Dans ces deux techniques, en cas d’hypertélorisme très important ou lorsqu’il existe une peau dystrophique ou très inesthétique, une résection cutanée centrofaciale est réalisée (fig. 6-18). Les canthi doivent systématiquement être repositionnés par une canthopexie transnasale. Si nécessaire, les gestes sur les voies lacrymales peuvent être réalisés dans le même temps.
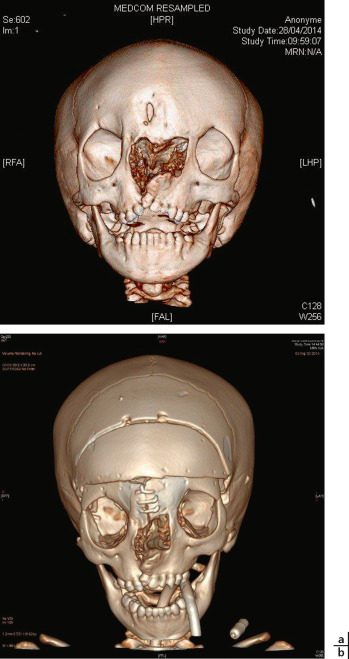
Fig. 6-27 Imagerie craniofaciale (TDM avec reconstruction tridimensionnelle) avant (a) et après (b) chirurgie de bipartition faciale pour corriger un hypertélorisme par fente faciale médiane et paramédiane droite (cas de l’enfant présenté à la figure 6-18).
Plusieurs techniques seront indiquées en fonction de la sévérité de l’atteinte des voies lacrymales. En cas de sténose, un sondage nasolacrymal, voire un sondage avec intubation peuvent être pratiqués précocement. En cas d’atteintes plus sévères, les gestes plus élaborés tels la dacryocystorhinostomie, la canaliculo-dacryocystorhinostomie sont généralement différés et effectués dans le même temps que la correction des bases osseuses.
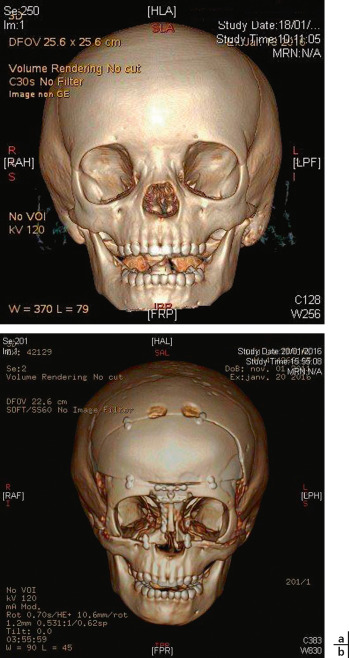
Fig. 6-28 Aspect en imagerie avant (a) et après (b) correction chirurgicale d’un hypertélorisme selon la technique des box (cas de l’enfant présenté à la figure 6-20).
La correction du ptosis est souvent réalisée en différé après correction des bases osseuses sauf en cas de risque majeur d’amblyopie à un âge où l’enfant peut être plus précisément examiné. Le geste est délicat chez le tout petit à cause du risque de surcorrection avec exposition cornéenne secondaire par non-occlusion ou malocclusion palpébrale.
Chez un très jeune enfant, les matériaux comme des bandes de silicone ou de polytétrafluoroéthylène expansé (PTFEe) sont utilisés pour réaliser une suspension au muscle frontal.
À partir de 5 ans, des techniques de résection ou plicature du muscle releveur peuvent être utilisées ou encore de suspension au muscle frontal par matériau autologue (par exemple aponévrose temporale ou tenseur du fascia lata).
Parce que la chirurgie craniofaciale principale peut améliorer ou aggraver un problème palpébral, on essaie aussi souvent que possible de différer la prise en charge palpébrale après cette chirurgie. La réparation du ptosis est plus volontiers différée jusqu’à ce qu’une chirurgie définitive puisse être réalisée.
Les techniques chirurgicales [13] sont assez similaires quel que soit le type de fentes faciales rencontré. Dans les fentes faciales latérales (3, 4 et 5), il existe souvent un raccourcissement de la hauteur entre la paupière inférieure et la base alaire ou la lèvre supérieure. Les fentes osseuses sont corrigées après dissection large et désincarcération des parties molles par des greffes osseuses le plus souvent (au niveau du plancher orbitaire ou au niveau de la paroi antérieure du sinus maxillaire, par exemple) (fig. 6-29 à 6-31).
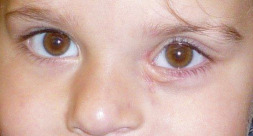
Fig. 6-29 Aspect postopératoire précoce après correction de la dystopie canthale interne par autoplastie locale – plastie de transposition palpébrale supérieure (cas de l’enfant présenté à la figure 6-22).
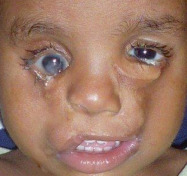
Fig. 6-30 Résultat postopératoire à 2 ans de la réparation bilatérale des fentes faciales obliques (cas de l’enfant présenté à la figure 6-23) comprenant des plasties locales avec lambeaux de rotation et d’avancée jugale et lambeau hétéropalpébral à gauche et multiples plasties étagées à droite.
La greffe orbitaire du plancher reste à prévoir : non réalisable lors de la première procédure : enfant extrêmement dénutrie et prise en charge en condition humanitaire.

Fig. 6-31 Aspect postopératoire à 3 ans de la correction chirurgicale de la dystopie canthale interne par lambeaux de transposition hétéropalpébraux et repositionnement du pied d’aile narinaire avec interposition d’une greffe composite auriculaire (cas de l’enfant présenté à la figure 6-17).
La région orbitopalpébrale est particulièrement concernée dans le cadre de malformations craniofaciales congénitales rares de type fente faciale. Le retentissement ophtalmologique doit être évalué au plus tôt et de la manière la plus précise possible. Celui-ci décide bien souvent du calendrier chirurgical. L’ophtalmologiste tient une place importante dans la décision chirurgicale. La correction des anomalies associe des techniques chirurgicales osseuses craniofaciales mais également des techniques de plastie cutanée pour la réparation des parties molles. La prise en charge de ces enfants nécessite une collaboration étroite entre les différentes spécialités réunies au sein de l’équipe de prise en charge des anomalies cranio- maxillo-faciales.
[1] Van der Meulen JC. Oblique facial clefts : pathology, etiology and reconstruction. Plast Reconstr Surg 1985 ; 76 : 212-24.
[2] Tessier P. Anatomical classification of facial, craniofacial and laterofacial clefts. J Maxillofac Surg 1976 ; 4 : 69-92.
[3] Van der Meulen JC, Mazzola R, Stricker M, Raphael B. Classification of craniofacial malformations. In : Stricker M, Van der Meulen JC, Raphael B, et al. Eds. Craniofacial malformations. Edimbourg : Churchill Livingston ; 1990, p. 149-309.
[4] Mazzola RF, Mazzola IC. Facial clefts and facial dysplasia : revisiting the classification. J Craniofac Surg 2014 ; 25 : 26-34.
[5] Fries PD, Katowitz JA. Congenital craniofacial anomalies of ophthalmic importance. Surv Ophthalmol 1999 ; 35 : 87-119.
[6] Heinrich A, Proff P, Michel T, et al. The kaleidoscopic world of rare craniofacial clefts: order out of chaos (Tessier classification). Clin Plast Surg 1976 ; 3 : 529e572.
[7] Ortiz-Monasterio F, Molina F. Orbital hypertelorism. Clin Plast Surg 1994 ; 21 : 599-612.
[8] Van den Elzen ME, Versnel SL, Wolvius EB, et al. Long-term results after 40 years experience with treatment of rare facial clefts : part 2 – symmetrical median clefts. J Plast Reconstr Aesthet Surg 2011 ; 64 : 1344-52.
[9] Alonso N, Freitas Rda S, de Oliveira E, et al. Tessier no. 4 facial cleft. Evolution of surgical treatment in a large series of patients. Plast Reconstr Surg 2008 ; 122 : 1505-13.
[10] Resnick JI, Kawamoto Jr HK. Rare craniofacial clefts : Tessier no. 4 clefts. Plast Reconstr Surg 1990 ; 85 : 843-9.
[11] Boo-Chai K. The oblique facial cleft : a 20-year follow-up. Br J Plast Surg 1990 ; 43 : 355-8.
[12] Genevieve D, Captier G, Blanchet C. Syndromes avec fentes labiopalatines. In : Lacombe D, Philip N. Syndromes dysmorphiques. Doin ; 2013, p. 271-4.
[13] Poswillo D. The pathogenesis of the first and second branchial arch syndrome. Oral Surg Oral Med Oral Pathol 1973 ; 35 : 302-28.
[14] Kaban LB, Padwa B, Mulliken JB. Mandibular deformity in hemifacial microsomia : a reassessment of the Pruzansky and Kaban classification. Plast Reconstr Surg 2014 ; 134 : 657-8.
[15] Mansour AM, Wang F, Henkind P, et al. Ocular findings in the facioauriculovertebral sequence (Goldenhar-Gorlin syndrome). Am J Ophtalmol 1985 ; 100 : 555-9.
[16] Baum JL, Feingold M. Ocular aspects of Goldenhar’s syndrome. Am J Ophthalmol 1973 ; 75 : 250-7.
[17] Mishra S, Sabhlok S, Panda PK, Khatri I. Management of midline facial clefts. J Maxillofac Oral Surg 2015 ; 14 : 883-90.
E. Zanin et C. Benso-Layoun, A. Aziz-Alessi , D. Denis
Les anomalies développementales du globe oculaire (ADG) sont des pathologies rares aboutissant à un globe de petite taille. Ces ADG sont un signe d’appel important devant faire redouter une malformation oculaire et/ou systémique (associées dans un tiers des cas). La sévérité de ces anomalies est variable allant de la simple asymétrie de taille des globes à une anophtalmie en fonction de l’arrêt plus ou moins précoce du développement de l’ébauche oculaire. Elles regroupent l’anophtalmie, la microphtalmie postérieure simple, la microphtalmie complexe et la nanophtalmie. Cette dernière présente des caractéristiques très spécifiques. L’incidence de l’anophtalmie et de la microphtalmie est respectivement de 2 et 19 pour 10 000 naissances vivantes [1]. Le diagnostic peut être anténatal grâce aux échographies systémiques de dépistage complétées en cas de doute par une IRM cérébro-orbitaire foetale ou néonatale. La prise en charge des ADG pose plusieurs problèmes [2] :
recherche de l’étiologie (génétique, embryopathie) ;
gestion du contexte polymalformatif (un tiers des cas) ;
pronostic fonctionnel variable en fonction du degré d’atteinte (uni- ou bilatérale, pure ou complexe) ;
prise en charge de la cataracte lorsqu’elle est associée dans les cas de microphtalmies complexes ;
prise en charge de la dilatation de la cavité et des culs-de-sac par conformateurs de tailles croissantes afin d’assurer la croissance correcte du cadre orbitaire ;
prise en charge esthétique de la micro-orbitie.
Le diagnostic d’ADG est avant tout clinique devant une diminution visible du volume du globe. Celui-ci peut être aisé en cas de microcornée associée ou d’asymétrie de taille entre les deux globes. Cependant, en cas de microphtalmie postérieure simple bilatérale, il sera retardé car sans point d’appel visible. Dans tous les cas, le diagnostic devra être confirmé par des mesures biométriques objectives réalisées lors d’un examen sous anesthésie générale comprenant : les diamètres cornéens au compas de blanc à blanc (microcornée si < 10 mm à la naissance et < 11 mm à l’âge adulte), la kératométrie, la longueur axiale, la profondeur de la chambre antérieure, l’épaisseur du cristallin. Un examen ophtalmologique complet sera également réalisé avec une réfraction sous cycloplégie, un examen à la lampe à fente du segment antérieur, un FO. La rétine devra bénéficier d’une analyse structurale et fonctionnelle par OCT et électrorétinogramme (ERG) à adapter à l’âge de l’enfant. Au décours de ce bilan, l’ADG sera classée en anophtalmie, microphtalmie postérieure simple, microphtalmie complexe ou nanophtalmie. Ce bilan ophtalmologique devra être associé à un examen pédiatrique et une imagerie cérébro-orbitaire (fig. 6-32) à la recherche de malformations systémiques associées. Il sera important d’examiner les parents et la fratrie et d’adresser l’enfant aux généticiens afin d’évaluer le risque de transmission.
L’anophtalmie est l’absence totale du ou des globes oculaires. Cette pathologie est très rare et sa cause est l’échec complet du développement de la vésicule optique primaire [3]. Il existe quatre types d’anophtalmie [4] :
primaire : absence totale d’ébauche oculaire et de voie optique. Il n’y a pas d’anomalie du tube neural. L’atteinte est bilatérale, sporadique, sans anomalie associée. Le trouble se produirait dès la fin de la première semaine ;
secondaire : il existe une anomalie de la partie antérieure du tube neural et le foetus est non viable ;
avec oeil kystique par non-invagination de la vésicule optique. La taille de l’oeil kystique est variable et peut croître après la naissance. Le défaut d’organogenèse aurait lieu entre 3 et 4 semaines de grossesse ;
consécutive ou dégénérative : la vésicule optique s’est formée mais a dégénéré secondairement. Elle correspond à l’anophtalmie clinique ou la microphtalmie extrême. Cette forme est très rare. Le trouble aurait lieu vers la 5e semaine de grossesse.
Les patients anophtalmes ont une incidence élevée d’anomalies développementales cérébrales et systémiques [4].
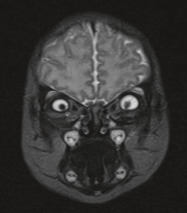
Fig. 6-32 IRM cérébro-orbitaire d’un nouveau-né présentant une microphtalmie sévère bilatérale, séquence T2 TSE, coupe coronale.
La microphtalmie postérieure correspond à un oeil sans malformation, avec un segment antérieur normal et une longueur axiale (LA) faible. En effet, le segment antérieur ne présente aucune anomalie malformative visible à la lampe à fente et sa biométrie est normale : diamètres cornéens (DC), profondeur de chambre antérieure (CA) et épaisseur du cristallin [3, 5]. Les valeurs de LA retenues pour le diagnostic de microphtalmie sont à corréler à l’âge : diminution de 2 DS par rapport à la moyenne des individus du même âge soit : < 14 mm chez un nouveau-né, < 19 mm à 1 an [3], < 20,5 mm chez l’adulte [5]. Les autres paramètres biométriques du segment antérieur sont normaux : DC horizontal ≥ 11 mm, profondeur de CA entre 3,14 et 3,6 mm, épaisseur du cristallin entre 4 et 4,45 mm [5]. L’hypermétropie est comprise entre 7 et 18 D. L’angle iridocornéen est dans la majorité des cas ouvert et le risque de fermeture de l’angle moins important que pour les nanophtalmies. Les microphtalmies les plus sévères peuvent présenter un pli rétinien inter-papillo-maculaire (24 % des cas) [5, 6]. On peut noter plus rarement d’autres anomalies rétiniennes telles que : stries fines rétiniennes, rétinite pigmentaire, pseudo-oedème papillaire et trous maculaires. L’acuité visuelle est variable selon l’importance de la microphtalmie et l’atteinte rétinienne. De multiples facteurs sont incriminés dans la genèse de la microphtalmie postérieure simple : une taille anormale du disque optique, une pression oculaire anormalement basse, un vitré altéré, une production anormale d’hormone de croissance. La microphtalmie peut être associée avec des anomalies systémiques développementales dans environ 50 % des cas (syndrome d’alcoolisation foetale, embryopathie diabétique, achondroplasie, mucopolysaccharidose, etc.).
La microphtalmie complexe est caractérisée par l’association d’une microphtalmie et de malformations développementales. Les anomalies oculaires peuvent être colobomateuses (fig. 6-33) ou non [4] et concerner le segment antérieur (sclérocornée, cataracte, etc.) ou postérieur (persistance du vitré primitif, dysplasie rétinienne, etc.). Des formes sporadiques ou héréditaires peuvent se rencontrer. Les facteurs environnementaux sont fortement suspectés dans la survenue des formes sporadiques. L’expansion du vitré étant responsable de 60 % de la croissance du segment postérieur en prénatal et 90 % en postnatal, une production anormale du vitré secondaire pourrait être à l’origine de ces microphtalmies complexes avec atteinte du segment postérieur [4]. L’acuité est variable allant d’une acuité relativement conservée à l’absence de perception visuelle. Dans ce groupe, on peut identifier des formes particulières :
microphtalmie complexe avec colobome qui est secondaire à un défaut de fermeture de la fissure embryonnaire au cours de la 7e semaine. Le colobome peut concerner l’iris, le cristallin, la choriorétine ou le nerf optique. La forme bilatérale autosomique dominante sans anomalie systémique associée est la plus fréquente [4] ;
microphtalmie complexe avec kyste : c’est une malformation colobomateuse majeure qui résulte d’un défaut de fermeture de la fissure embryologique et se caractérise par l’existence d’une masse orbitaire associée à un oeil microphtalme avec des malformations sévères. La taille du kyste peut être très variable. La majorité des cas sont sporadiques, unilatéraux et sans malformation systémique associée. Certaines formes enveloppent le globe le rendant invisible et font craindre une tumeur orbitaire ;
microphtalmie avec persistance du vitré primitif (fig. 6-34). Elle est unilatérale lorsqu’elle est isolée et bilatérale lorsqu’elle est syndromique. On note au niveau du segment antérieur une cataracte postérieure ou totale. Le réseau vasculaire peut être visible à la lampe à fente, il part en corolle à partir de l’artère hyaloïdienne le long de la capsule postérieure et peut s’étendre à la capsule antérieure. On peut voir une poussée postérieure avec athalamie. Seules les artères hyaloïdiennes de gros calibre sont visibles en échographie en mode B.
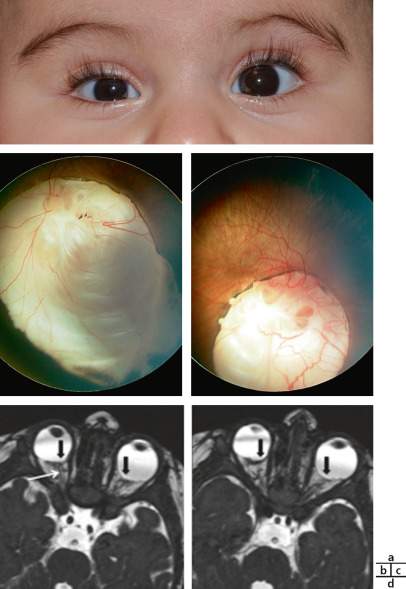
Fig. 6-33 Nourrisson de 8 mois présentant une microphtalmie droite (a).
b, c. Yeux droit (b) et gauche (c). Photographies du fond d’oeil à la RetCam™ : colobome papillo-chorio-rétinien bilatéral ; plus sévère à droite où le colobome englobe la papille et une large partie du pôle postérieur en inférieur avec absence de tissu rétinien et choroïdien en regard. d. IRM cérébro-orbitaire, séquence T2, coupe axiale passant par les nerfs optiques, montrant le colobome papillo-chorio-rétinien bilatéral plus vaste à droite et une atrophie sévère du nerf optique droit (flèche).
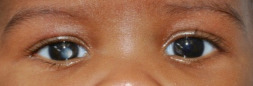
Fig. 6-34 Microphtalmie droite minime avec persistance de vitré primitif chez un nourrisson de 4 mois.
Néanmoins, le fond d’oeil étant souvent inaccessible, une échographie en mode B sera indispensable afin d’évaluer l’état rétinien ;
microphtalmie et dysplasie rétinienne : association très fréquente. Elle peut être mineure et périphérique ou majeure avec un décollement de rétine total ;
cryptophtalmie et microphtalmie : atteinte rare caractérisée par l’absence de fente palpébrale (aucune solution de continuité). Cette malformation est fréquemment associée à des anomalies oculaires (dysgénésie du segment antérieur, opacité cornéenne, insuffisance limbique, etc.), faciales (déformation nasale, fentes labiales et/ou palatine), auriculaires (oreille moyenne et externe), rénales (agénésie), génitales, squelettiques (syndactylies devant faire évoquer le syndrome de Fraser : fig. 6-35), et neurologiques (retard mental).
Les microphtalmies complexes sont fréquemment associées à des atteintes systémiques rentrant dans le cadre d’associations syndromiques. Il en existe plus de 200 (Tableau 6-6).
Tableau 6-6 – Principaux syndromes associés à une anomalie développementale du globe oculaire (ADG).
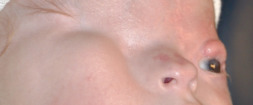
Fig. 6-35 Nouveau-né de 3 mois atteint de syndrome de Fraser.
Cryptophtalmie droite, colobome de la paupière supérieure gauche ayant bénéficié d’un lambeau de reconstruction frontal pédiculé, absence de culde-sac conjonctival supérieur, insuffisance limbique sur 360° et ulcère cornéen.
C’est une forme extrême, rare et spécifique de microphtalmie. On note un arrêt du développement du globe dans toutes ses dimensions après la fermeture de la fissure embryonnaire. Elle est généralement bilatérale et non associée à des malformations oculaires ou systémiques. Les rares formes unilatérales peuvent être associées à une hypoplasie de l’hémiface ou de l’hémicorps correspondant. L’expansion du globe devient impossible par une anomalie de composition de la sclère qui la rend épaisse, inélastique et précocement fibrosée avec l’âge entraînant une compression des veines vortiqueuses avec obstacle au retour veineux. Les patients atteints présentent un faciès caractéristique avec des fentes palpébrales étroites et des petits yeux enfoncés dans les orbites (fig. 6-36).
La nanophtalmie est caractérisée par : une hypermétropie forte strictement supérieure à 7 D qui peut aller jusqu’à 17-18 D (fig. 6-37) ; une longueur axiale très courte (< 20,5 mm chez l’adulte) ; une microcornée (diamètre cornéen entre 9,5 et 11 mm) [2, 5] ; un cristallin épais ; une chambre antérieure étroite ; une sclère épaisse (5 mm) et inélastique avec des anomalies du collagène et des protéoglycannes [7]. Rehlan et al. exposent dans une série de 38 patients forts hypermétropes les différentes caractéristiques entre microphtalmie et nanophtalmie (Tableau 6-7) [5]. Le rapport volume du cristallin par rapport à l’oeil est 4 à 8 fois supérieur à la normale, ce qui entraîne une chambre antérieure étroite, une convexité irienne, une fermeture progressive de l’angle iridocornéen avec survenue d’un glaucome par fermeture de l’angle entre 40 et 60 ans. On peut noter au FO ainsi qu’à l’OCT des dépôts jaunâtres fovéolaires, une hypoplasie fovéolaire (fig. 6-38), des stries rétiniennes, un oedème maculaire en rapport avec une rétinite pigmentaire évoluée ou, plus rarement que pour les microphtalmies postérieures simples, un pli rétinien interpapillo- maculaire [8].
La nanophtalmie est fréquemment associée à une rétinite pigmentaire (39 % selon Relhan et al. [5]), elle doit donc être recherchée de manière systématique par la réalisation d’un ERG. Les autres anomalies décrites sont, au niveau ophtalmologique, des drusen du nerf optique, et au niveau systémique, les syndromes de Weil-Marchesani, de Hallerman-Streiff, de Kenny-Caffey, etc. (Tableau 6-6). La majorité des cas sont sporadiques. On retrouve une hérédité avec des formes dominantes : atteinte des chromosomes 11p (NNO1), 2q11-q14 (NNO3) (fig. 6-39) et des formes récessives liées à des mutations du gène MFRP au niveau du chromosome 11q23.
Chez l’enfant, l’acuité visuelle corrigée est correcte et semble meilleure que dans la microphtalmie postérieure [5] en dehors des cas d’hypoplasie fovéolaire associée. Avec l’âge, les yeux évoluent inexorablement vers un glaucome par fermeture de l’angle dont la prise en charge expose à des complications redoutables. La fermeture de l’angle survient habituellement entre 20 et 50 ans mais peut se voir dès l’enfance. Les anomalies de structure et d’épaisseur sclérale diminuent sa perméabilité provoquant une accumulation de liquide dans l’espace suprachoroïdien (fig. 6-40). Il en résulte un gradient de pression postérieur avec une expansion choroïdienne et une rotation antérieure des corps ciliaires, pouvant être à l’origine d’une effusion uvéale aiguë spontanée (décollements choroïdiens et décollements séreux rétiniens) ou chronique (altérations de l’épithélium pigmentaire visible au FO et aux clichés en autofluorescence). Les risques sont majorés après un laser pour iridotomie et maximaux après une chirurgie de cataracte ou de glaucome pouvant entraîner effusion uvéale, glaucome malin, décollement de rétine non rhegmatogène de prise en charge très complexe ou hémorragie expulsive. Le suivi et le traitement sont complexes (encadré 6-2). En fonction du degré de fermeture de l’angle et de la pression intra-oculaire (PIO), la prise en charge thérapeutique comprendra : iridotomie ± iridoplastie laser, collyres hypotonisants, chirurgie filtrante, extraction cristallinienne ou cyclo-affaiblissement par laser diode. Dans tous les cas, il faudra prévenir le patient de la complexité des gestes et des risques encourus. Une surveillance précoce et régulière de la PIO et de la gonioscopie est nécessaire. Dès que l’angle devient étroit, même avec une PIO normale, on pourra proposer une iridotomie laser (difficile techniquement en raison de l’épaisseur de l’iris et de la proximité de la cornée), puis une iridoplastie laser périphérique si l’iridotomie est insuffisante pour prévenir les goniosynéchies. Si la PIO augmente, il faudra privilégier le traitement médical par collyres hypotonisants. En cas d’échec, une chirurgie filtrante sera indiquée. Il faudra préférer la sclérectomie profonde non perforante en première intention si l’angle iridocornéen est non synéchié (rare). En cas de goniosynéchies, une trabéculectomie devra être réalisée avec les précautions suivantes : réalisation de sclérotomies ou de trappes sclérales prophylactiques [7] (2 à 3 semaines avant la chirurgie filtrante ou en peropératoire) afin d’améliorer la perméabilité et l’élasticité sclérale, utilisation de visco-élastique pour éviter une décompression brutale et sutures préplacées pour éviter l’hémorragie expulsive. En cas de glaucome malin, il faudra réaliser une vitrectomie en urgence. L’extraction du cristallin permet une augmentation globale du volume de la chambre antérieure, une amélioration de l’acuité visuelle dans la majorité des cas et une réduction de la PIO [9]. La biométrie est difficile en raison de la grande variation de profondeur de chambre antérieure entre les mesures pré- et postopératoires. Les risques et difficultés peropératoires sont : une mauvaise dilatation, un iridoschisis, une rupture capsulaire postérieure, la rigidité des implants à forte puissance voire la nécessité d’utiliser un piggy-back. Les complications postopératoires sont : oedème cornéen, réaction inflammatoire, oedème maculaire cystoïde, effusion uvéale, glaucome malin et opacification capsulaire secondaire. Pour les nanophtalmies majeures, il est conseillé de réaliser des sclérotomies ou trappes sclérales prophylactiques en peropératoire. Ces incisions sont également proposées pour le traitement des effusions uvéales spontanées ou secondaires.

Fig. 6-36 Enfant de 6 ans atteint de nanophtalmie bilatérale, faciès particulier avec fentes palpébrales étroites et globes oculaires enfoncés dans les orbites.
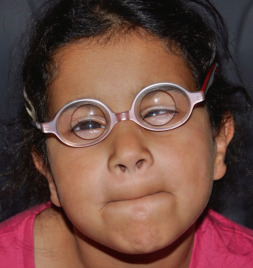
Fig. 6-37 Correction optique de nanophtalmie, réfraction sous cycloplégie.
OEil droit : + 18,75 (+ 1 à 50°). OEil gauche : + 19 (+ 0,50 à 100°).
Tableau 6-7 – Tableau récapitulatif des résultats de l’étude de Rehlan et al. [5] sur la comparaison des caractéristiques cliniques et biométriques des microphtalmies postérieures simples et des nanophtalmies*.
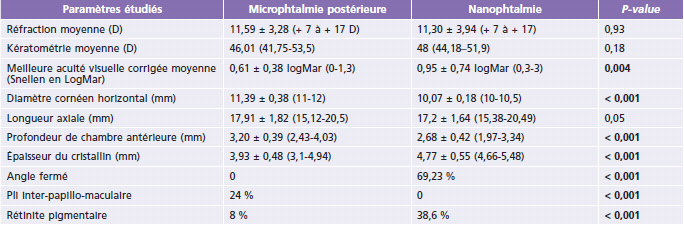
Fig. 6-38 OCT et fond d’oeil d’une enfant nanophtalme avec fovéa plana.
a. OEil droit. b. OEil gauche.

Fig. 6-39 Mère et fille nanophtalmes bilatérales.
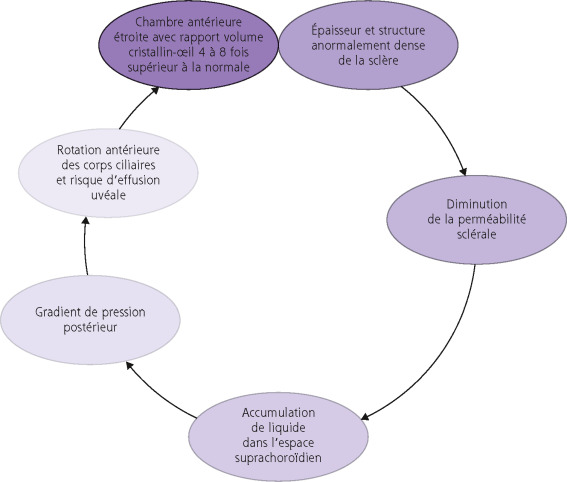
Fig. 6-40 Cercle vicieux de la nanophtalmie.
Prise en charge des nanophtalmies
Surveillance précoce et régulière de la pression intraoculaire et de la gonioscopie.
Iridotomie et iridoplastie lasers précoces avant la survenue d’une hypertonie pour éviter les goniosynéchies.
En cas d’hypertonie oculaire, privilégier le traitement médical.
Si chirurgie filtrante nécessaire : sclérectomie si angle, ouvert sinon trabéculectomie avec précautions.
Phacoémulsification bénéfique sur la profondeur de chambre, la pression intra-oculaire et l’acuité visuelle, mais risques opératoires importants.
Toujours mettre en balance les bénéfices et les risques.
Les causes d’anophtalmie et de microphtalmie sont complexes et multiples. On distingue les causes génétiques/chromosomiques, et les embryofoetopathies par infection ou exposition à des facteurs environnementaux.
De 23 à 30 % des ADG sont de cause chromosomique. Un grand nombre de délétions, duplications, translocations ont été reliées à l’anophtalmie et à la microphtalmie. Plusieurs gènes ont été identifiés : SOX2 (principal gène à l’origine de microphtalmie et anophtalmie avec hérédité autosomique dominante), PAX6, PAX2, CHX10, FOXE3, OTX2, BMP4 [3]. La trisomie 13 est particulièrement pourvoyeuse de microphtalmie complexe (colobome, dysplasie rétinienne). Elle est caractérisée par un faciès particulier (microcéphalie, menton fuyant, oreilles basses, bec-de-lièvre) des doigts surnuméraires (60 à 80 % ) et une aplasie du vertex en « aile de papillon » . La trisomie 18 entraîne une atteinte ophtalmologique moins sévère avec microphtalmie modérée, opacités cornéennes (5 à 15 % ), colobome du nerf optique, anomalies orbitopalpébrales (hypertélorisme, épicanthus, ptosis). En général, il s’agit d’enfant de sexe féminin, né de mère âgée avec une dysmorphie craniofaciale, un chevauchement de l’index sur le médius et une cardiopathie limitant leur espérance de vie [2].
Les infections prénatales virales (rubéole, varicelle, cytomégalovirus, herpès, parvovirus B19, coxsackies) ou parasitaires (toxoplasmose) sont incriminées dans la survenue des anomalies développementales du globe oculaire.
La carence maternelle en vitamine A, l’exposition aux rayons X, l’exposition aux antimitotiques, la prise de toxiques (thalidomide, alcool, warfarine, solvant, LSD) sont des causes d’ADG [2].
Les objectifs thérapeutiques sont une optimisation des capacités visuelles et la gestion de l’aspect esthétique en stimulant la croissance des tissus mous et des cavités orbitaires osseuses. Trois problématiques se posent.
La question de la correction du vice de réfraction se pose en particulier en cas de microphtalmie postérieure simple ou de nanophtalmie (hypermétropie forte). Cette prise en charge sera d’autant plus importante que l’atteinte est unilatérale ou asymétrique avec une anisométropie conséquente. La correction devra être totale, précoce à port permanent. Un traitement d’amblyopie par occlusion sera nécessaire.
L’association microphtalmie-cataracte est fréquente. La cataracte devra être prise en charge chirurgicalement à 6 semaines en cas d’atteinte unilatérale et à 2-3 mois en cas d’atteinte bilatérale et en fonction des anomalies systémiques associées. Un bilan préopératoire avec réalisation d’une échographie en mode B à la recherche d’une persistance du vitré primitif et un bilan pédiatrique complet à la recherche d’une malformation systémique associée (en particulier cardiaque) seront indispensables. Certaines notions devront être prises en compte [2] :
l’intervention est délicate sur un oeil microphtalme et l’implantation sera déconseillée précocement ;
le pronostic fonctionnel est le plus souvent médiocre, a fortiori lorsque la microphtalmie est sévère ;
l’amblyopie est inéluctable et profonde si l’atteinte est unilatérale et justifie une occlusion intense et précoce du bon oeil 80 % du temps d’éveil dès que l’oeil microphtalme opéré aura été équipé en lentilles rigides ;
un ERG sera nécessaire afin de dépister une dysplasie rétinienne.
Les ADG s’accompagnent d’une micro-orbitie, d’une hypoplasie du maxillaire homolatérale et d’une rétraction des culs-de-sac conjonctivaux. Il est capital de prendre en charge ce problème dès les premiers mois de vie par la mise en place de conformateurs plastiques de tailles croissantes. En effet, si la dilatation par conformateur n’est pas commencée assez tôt, il s’avérera impossible de placer une prothèse et il faudra réaliser une expansion chirurgicale de l’orbite. La prise d’empreinte des conformateurs est réalisée sous anesthésie générale et doit être renouvelée tous les 4 mois la première année afin d’adapter la taille des conformateurs à la croissance orbitaire (fig. 6-41 et fig. 6-42).
Dans le cas des microphtalmies sévères et des anophtalmies, le traitement reposera sur des stratégies de remodelage et de remplacement du volume endo-orbitaire (implant, ballonnet d’expansion, greffon dermo-graisseux) associées à la pose de conformateurs. Dans le cas de microphtalmie avec kyste, la prise en charge s’adaptera à la croissance orbitaire.
À noter qu’en cas d’atteinte bilatérale sévère, il est possible de prescrire de la mélatonine chez les enfants n’ayant pas de perception lumineuse afin de réguler le sommeil nocturne.
Fig. 6-41 Exemple d’adaptation en coque sclérale chez une enfant présentant une microphtalmie droite sévère.
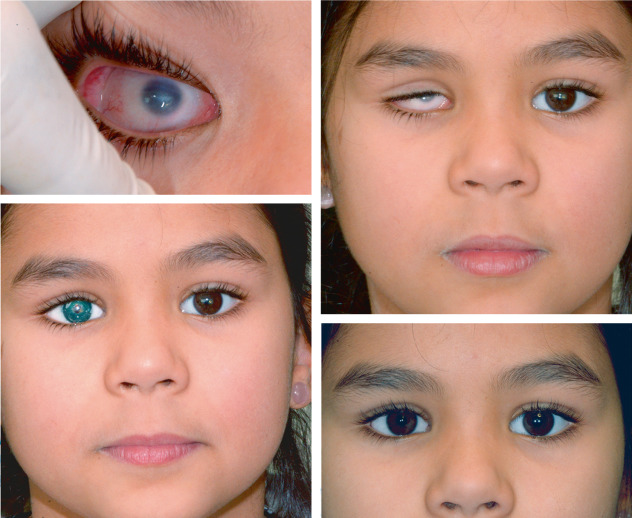
Fig. 6-42 Étapes de fabrication des conformateurs orbitaires.
Empreinte silicone ; maquette et contre-type ; réalisation de la prothèse.
Les ADG sont un symptôme d’importance majeure témoin d’une affection génétique ou d’une embryofoetopathie justifiant :
un bilan ophtalmologique et pédiatrique complet à la recherche de malformations associées et d’atteinte syndromique ;
une prise en charge multidisciplinaire précoce associant gestion des troubles réfractifs, de l’éventuelle cataracte associée, de l’amblyopie et de la croissance orbitaire par conformateurs ;
un conseil génétique afin de déterminer le risque pour la fratrie.
[1] Shaw GM, Carmicael SL, Yang W, et al. Epidemiologic characteristics of anophtalmia and bilateral microphtalmia among 2.5 million births in California 1989-1997. Am J Med Genet A 2005 ; 137 : 36-40.
[2] Godde Jolly D. Les malformations congénitales du globe oculaire. In : Godde Jolly D, Dufier JL. Ophtalmologie pédiatrique. Paris : Masson ; 1992, p. 75-83.
[3] Sachdeva R, Traboulsi E. Disorders of the eye as the whole. In : Taylor D, Hoyt CS. Pediatric ophthalmology and strabismus. 4th ed. London : Elsevier ; 2013, p. 139-46.
[4] Nischal KK. Anterior segment developmental anomalies. In : Wright KW, Ning Y, Strube J. Pediatric ophthalmology and strabismus. 3rd ed. Oxford university press ; 2015, p. 676-700.
[5] Relhan N, Jalali S, Pehre N, et al. High-hyperopia database, part I : clinical characterisation including morphometric (biometric) differentiation of posterior microphthalmos from nanophthalmos. Eye (Lond) 2016 ; 30 : 120-6.
[6] Park SH, Ahn YJ, Shin SY, Lee YC. Clinical features of posterior microphthalmos associated with papillomacular fold and high hyperopia. Clin Exp Optom 2016 ; 99 : 590-3.
[7] Jin JC, Anderson DR. Laser and unsutured sclerotomy in nanopthalmos. Am J Ophthalmol 1990 ; 109 : 575-80.
[8] Helvacioglu F, Kapran Z, Sencan S, et al. Optical coherence tomography of bilateral nanophthalmos with macular folds and high hyperopia. Case Reports in Ophthalmological Medicine 2014 ; 2014 : 173853.
[9] Ye Z, Li Z, He S, et al. Outcomes of coaxial micro-incision phacoemulsification in nanophthalmic eyes : report of retrospective case series. Eye Sci 2015 ; 30 : 94-100.
L. Desjardins
Les tumeurs orbitaires pédiatriques sont des pathologies peu fréquentes d’étiologies très variées, certaines étant propres à l’enfant. Les deux plus importantes séries publiées dans la littérature sont celles de Kodsi et de Shields avec respectivement 340 tumeurs orbitaires pédiatriques sur une période de 60 ans, et 285 sur 30 ans [1, 2]. Ces tumeurs sont bénignes dans plus de 80 % des cas, les plus fréquentes étant les tumeurs vasculaires (lymphangiome et angiome capillaire), les kystes dermoïdes et le gliome du nerf optique. Les tumeurs malignes, plus rares, sont essentiellement représentées par le rhabdomyosarcome et les localisations secondaires (neuroblastome métastatique, leucémies).
Le symptôme prédominant est en général l’exophtalmie. L’interrogatoire et l’examen clinique doivent être soigneux. On éliminera les fausses exophtalmies (myopie forte unilatérale, buphtalmie, glaucome congénital). L’examen des paupières peut parfois orienter le diagnostic : présence d’un hémangiome capillaire cutané ou d’une coloration bleutée évoquant un hémangiome sous-cutané. La présence de signes inflammatoires peut faire évoquer un rhabdomyosarcome, une lésion infectieuse (cellulite orbitaire), ou une pathologie inflammatoire. La palpation du cadre orbitaire permet de rechercher une masse, d’apprécier sa consistance et sa localisation. L’acuité visuelle et l’examen du fond d’oeil, à la recherche d’un oedème papillaire, d’une atrophie optique ou de plis choroïdiens, compléteront systématiquement le bilan ophtalmologique.
Les antécédents doivent être détaillés (maladie de Von Recklinghausen, traumatisme récent). L’âge de début, l’uni- ou la bilatéralité de l’exophtalmie sont des éléments très importants. Avant 2 ans, l’hémangiome capillaire ou le kyste dermoïde sont les causes les plus fréquentes. La vitesse d’évolution des symptômes est aussi un élément capital. Des tumeurs bénignes telles que l’hémangiome ou le kyste dermoïde grossissent lentement, alors qu’une exophtalmie augmentant rapidement suggère plutôt une tumeur métastatique ou un rhabdomyosarcome. En pratique, toute exophtalmie unilatérale rapidement évolutive doit être considérée jusqu’à preuve du contraire comme un rhabdomyosarcome, et nécessite la réalisation d’une imagerie orbitaire et d’une biopsie en urgence.
Une exophtalmie bilatérale rapidement évolutive et s’accompagnant d’une ecchymose périorbitaire est habituellement en rapport avec un neuroblastome métastatique. Dans la cellulite orbitaire, l’exophtalmie peut être également assez brutale mais elle s’accompagne souvent de douleurs, d’une limitation importante de mouvements oculaires et de fièvre avec altération de l’état général. Une altération de l’état général est aussi retrouvée dans les leucémies qui peuvent être à l’origine d’un sarcome granulocytique de l’orbite. L’examen général ne doit pas être négligé. Des taches café au lait peuvent suggérer la présence d’une neurofibromatose de Von Recklinghausen. Des lésions cutanées peuvent également être trouvées dans l’histiocytose, le xanthogranulome juvénile et l’hémangiome capillaire. Enfin, lorsque l’on suspecte un problème métastatique, la palpation et l’échographie de l’abdomen peuvent facilement orienter le diagnostic en retrouvant une masse abdominale.
Les examens complémentaires sont l’échographie orbitaire, le scanner, l’IRM et l’examen histologique.
L’échographie orbitaire est une technique non invasive, ne nécessitant aucune préparation, et facilement accessible. Elle permet souvent d’orienter le diagnostic et doit être recommandée en première intention. Elle permet de détecter une masse orbitaire, d’évaluer sa nature liquidienne ou solide, d’apprécier sa vascularisation en mode Doppler et de guider éventuellement une cytoponction ou une biopsie à visée diagnostique.
Le scanner permet d’analyser parfaitement les parois osseuses de l’orbite et son contenu, d’analyser les masses tumorales, leur extension dans l’orbite et leur rapport avec les structures extraet intraconiques. L’avantage du scanner par rapport à l’IRM est sa meilleure disponibilité et sa qualité relativement reproductible sur les machines multicoupes actuelles. Néanmoins, sauf urgence, il est aujourd’hui recommandé de réaliser une IRM en première intention chez l’enfant, car le scanner est une technique exposant aux radiations ionisantes pouvant favoriser la survenue de lésions malignes secondaires.
L’IRM est un examen plus long, moins facilement disponible, et nécessitant une sédation efficace voire une anesthésie générale chez les jeunes enfants. L’examen doit être réalisé en antenne crâne pour analyser non seulement la lésion orbitaire, mais aussi ses éventuelles extensions au massif facial, aux cavités sinusiennes et possiblement intracrâniennes.
Parfois, le diagnostic est suffisamment évident sur les critères clinico-radiologiques pour que l’on se passe de la biopsie : c’est le cas pour les tumeurs vasculaires bénignes par exemple (hémangiomes du nourrisson, lymphangiome). Mais dans la plupart des cas, le diagnostic étiologique suspecté sur les éléments cliniques et radiologiques, repose sur la biopsie avec examen histologique. Lorsque l’on réalise cette biopsie, il faut toujours penser à garder du tissu frais pour d’éventuelles études en biologie moléculaire. Si l’on suspecte un neuroblastome métastatique, la biopsie doit être réalisée au niveau de la tumeur primitive. En cas de métastase, de sarcome granulocytique ou de lymphome, un examen cytologique peut parfois être très utile [3].
Les tumeurs orbitaires pédiatriques peuvent être classées selon différents critères : fréquence, âge de survenue, caractère acquis ou congénital, bénin ou malin. Nous avons opté ici pour une classification principalement fondée sur l’origine tissulaire, classification déjà utilisée pour le rapport SFO 1998 (J.-P. Adenis et S. Morax). Cette classification garde quelques imperfections, les lésions histiocytaires pouvant par exemple être classées dans les lésions lymphoprolifératives comme dans les tumeurs osseuses.
Le rhabdomyosarcome est une tumeur maligne à différenciation musculaire striée. C’est la tumeur orbitaire maligne primitive la plus fréquente chez l’enfant. Cette tumeur d’évolution rapide est une urgence thérapeutique. L’âge moyen au diagnostic est de 7 à 8 ans. L’exophtalmie, très rapidement évolutive, est retrouvée dans 71 % des cas (fig. 6-43a et b). Elle peut s’accompagner souvent de signes inflammatoires et d’un gonflement des paupières (fig. 6-43c). Une masse palpable est présente dans 58 % des cas. Une localisation palpébrale peut parfois orienter à tort au départ vers un chalazion (fig. 6-44). Dans la majorité des cas, il s’agit d’une maladie à développement exclusivement orbitaire, les atteintes à distance ganglionnaires, prétragiennes ou cervicales, ou métastatiques étant exceptionnelles (< 2 % ).
L’imagerie doit être réalisée très rapidement. Elle permet de réaliser le bilan d’extension initial et de guider la biopsie. Le scanner ne reste indiqué qu’en cas de défaut d’accès à l’IRM dans un délai rapide. L’existence d’une extension « paraméningée » de grande valeur pronostique doit être recherchée, celle-ci étant définie par un envahissement du toit de l’orbite, de la fente sphénoïdale, une tumeur au contact du nerf optique ou un envahissement des sinus ethmoïdaux ou maxillaires [4].
Le diagnostic de certitude repose sur la biopsie chirurgicale de la tumeur avec analyse histologique et en biologie moléculaire. Ces analyses seront confiées de préférence à un anatomopathologiste spécialisé. Une partie du matériel tumoral doit systématiquement être congelée (tumorothèque), afin de pouvoir pratiquer des analyses biologiques complémentaires si celles-ci s’avèrent nécessaires secondairement. Un marquage par la desmine peut être utile. L’histologie permet de classer les rhabdomyosarcomes en deux sous-types, embryonnaire et alvéolaire. Le rhabdomyosarcome embryonnaire est plus fréquent, environ 80 % des cas, et de meilleur pronostic que le rhabdomyosarcome alvéolaire, avec une survie respectivement de 94 % et 74 % . Certaines anomalies génétiques pathognomoniques permettent de poser formellement le diagnostic du sous-type alvéolaire de rhabdomyosarcome. Les plus fréquentes sont les translocations chromosomiques (1;13) et (2;13) correspondant aux transcrits de fusion PAX3/FKHR et PAX7/FKHR analysables en biologie moléculaire par reverse transcription-polymerase chain reaction (RT-PCR) ou par fluorescent in situ hybridization (FISH) [5].
Le traitement actuel ne se conçoit qu’au sein d’une équipe pluridisciplinaire. Il comprend de manière systématique une polychimiothérapie première de réduction tumorale. De nombreux médicaments sont efficaces, principalement l’ifosfamide, le cyclophosphamide, l’actinomycine, la vincristine et la doxorubicine. Afin d’améliorer le contrôle local de la maladie, le traitement systémique est souvent associé à un traitement local, réalisé en général après les premières cures de chimiothérapie. Dans la plupart des cas, le traitement local est une radiothérapie externe (36-50,4 Gy), en particulier lorsqu’il existe des facteurs de mauvais pronostic (âge supérieur à 10 ans, taille tumorale initiale supérieure à 5 cm, type histologique alvéolaire, extension paraméningée et/ou absence de rémission complète radiologique à l’issue des 3 premières cures de chimiothérapie) ou en cas de rechute. La technique d’irradiation est variable, irradiation externe, protonthérapie, certaines localisations palpébrales uniques pouvant aussi bénéficier d’une curiethérapie (prontothérapie : fig. 6-45 ; curiethérapie : fig. 6-46). Plus rarement, le traitement local consistera en une exérèse chirurgicale du résidu tumoral (petites tumeurs palpébrales en particulier). Du fait de la grande chimio/radio-sensibilité de cette tumeur, les chirurgies mutilantes, de type exentération, doivent être réservées uniquement aux tumeurs en rechute après radiothérapie [6].
Le pronostic des rhabdomyosarcomes orbitaires est excellent avec une survie sans événement à 5 ans de plus de 65 % et une survie globale de plus de 85 % . Les protocoles thérapeutiques actuels, tout en cherchant à maintenir cet excellent taux de survie, doivent s’astreindre à réduire les complications locales, notamment post-radiques (hypoplasie osseuse dans le territoire irradié, cataracte, sécheresse oculaire et parfois un déficit en hormone de croissance) [7], mais aussi générales liées à la chimiothérapie.
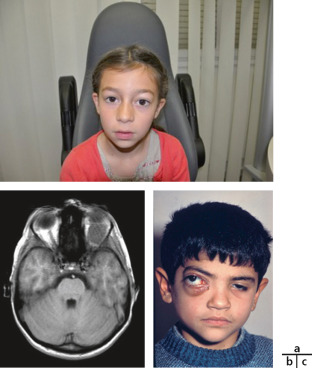
Fig. 6-43 Rhabdomyosarcomes.
a, b. Rhabdomyosarcome chez une enfant de 7 ans : tumeur révélée par une diplopie, une exophtalmie gauche (a) et développée aux dépens du droit supérieur gauche (b). c. Rhabdomyosarcome chez un enfant de 5 ans avec gonflement palpébral unilatéral droit, exophtalmie et exotropie.
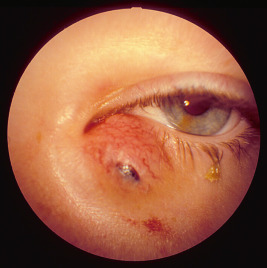
Fig. 6-44 Rhabdomyosarcome : localisation palpébrale.
Fig. 6-45 Protonthérapie d’une tumeur orbitaire.
Fig. 6-46 Curiethérapie d’une tumeur orbitaire.
L’hémangiome capillaire, ou hémangiome infantile bénin du nourrisson, est une tumeur vasculaire bénigne fréquente intéressant le plus souvent la peau des paupières et parfois la paupière et l’orbite ou l’orbite seulement. Elle est plus fréquente chez les filles. Elle est parfois présente dès la naissance ; mais dans deux tiers des cas, elle n’apparaît que quelques mois après la naissance. L’hémangiome a d’abord tendance à augmenter. La régression se fait ensuite spontanément, en général à partir de l’âge de 1 an (fig. 6-47). Environ 30 % des enfants présentent des angiomes cutanés typiques sur le corps (angiomes tubéreux). Le problème principal est le risque d’amblyopie fonctionnelle si la pupille est masquée, en cas d’anisométropie (en particulier en cas d’astigmatisme provoqué par un hémangiome antérieur), ou lorsqu’il existe une exophtalmie avec déviation du globe oculaire. Selon Schwartz [8], seuls les hémangiomes de diamètre supérieur à 1 cm sont associés à un risque d’amblyopie. Le diagnostic est confirmé par l’échographie-Doppler et/ou l’IRM qui retrouvent une lésion bien limitée, aux contours lobulés, contenant des vaisseaux artériels à flux élevé à basse résistance en Doppler. En IRM, la masse est de signal intermédiaire en T1, en hypersignal T2, et se rehausse de façon intense, précoce et homogène après injection de gadolinium. Sur le plan histologique, l’hémangiome capillaire est constituée de cellules endothéliales en mitose ayant tendance à former des espaces vasculaires à petite lumière irrégulière.
La prise en charge consiste à surveiller régulièrement pour dépister les problèmes d’amblyopie en attendant la régression spontanée qui est complète pour 60 % des patients à 4 ans et pour 76 % à 7 ans. En cas d’occlusion palpébrale ou d’augmentation rapide de la tumeur avec risque d’amblyopie, une thérapeutique plus active peut être envisagée. L’injection intralésionnelle de corticoïdes, en délivrant une haute concentration de cortisone au contact de l’hémangiome, permet une régression rapide du volume tumoral et une diminution de l’astigmatisme. Elle comporte néanmoins un risque d’occlusion de l’artère centrale (contrôle du FO après injection) et de nécrose de la paupière et n’est pas forcément dénuée d’effets secondaires généraux. Les injections sont en principe réservées aux formes superficielles. La corticothérapie par voie générale (prednisone 1 à 2 mg/kg/j) permet d’éviter les risques liés à l’anesthésie générale mais entraîne une régression plus lente et parfois des effets secondaires généraux. L’exérèse chirurgicale est utile pour les petites tumeurs localisées en particulier les formes avec amblyopie et résistant aux corticoïdes. Plus récemment, un traitement par bêtabloquant per os a été rapporté comme étant une alternative thérapeutique efficace et sans complication majeure. La dose habituellement utilisée de propranolol est de 2 mg/kg/j. Les effets secondaires connus du propranolol comportent un bronchospasme, une hypoglycémie, des perturbations de l’humeur et une somnolence, une bradycardie et une hypotension [9]. Ils sont réversibles à l’arrêt du traitement. Aucun cas de décès n’a été décrit. Le traitement serait efficace dans 96 % des cas.
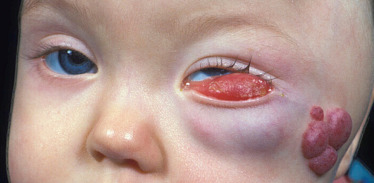
Fig. 6-47 Hémangiome.
Les lymphangiomes sont des malformations vasculaires infiltrantes composées de canaux entourés d’un endothélium dont l’aspect est celui des canaux lymphatiques. Ces hamartomes sont bénins, mais graves du fait d’un pronostic fonctionnel et esthétique réservé à long terme.
La plupart du temps, les symptômes débutent au cours des premières années de vie, bien que des débuts tardifs aient été décrits [10]. Dans beaucoup de cas, c’est à l’adolescence que le patient présente soudain une exophtalmie, un ptosis et une ecchymose traduisant un hématome spontané ou traumatique. Une composante conjonctivale, apparaissant sous la forme de kystes hémorragiques multiples (lymphangiectasies), est souvent présente et devrait suggérer l’existence d’un lymphangiome orbitaire associée. Ces formations kystiques remplies de sang (kystes chocolat) sont caractéristiques. L’hémorragie intralésionnelle brutale peut se résorber graduellement mais il existe un risque de récidive. La survenue des récidives hémorragiques peut être favorisée par des infections rhinopharyngée ou respiratoire. Souvent la maladie évolue ainsi par poussées successives et l’abord chirurgical lui-même est susceptible de provoquer une poussée évolutive. L’effet de masse induit par ces poussées évolutives peut entraîner une exophtalmie importante, une déviation du globe, une neuropathie optique compressive, un strabisme, une amblyopie et un préjudice esthétique. En imagerie, le lymphangiome apparaît comme une tumeur bien limitée se moulant autour des structures anatomiques, parfois hétérogène en cas de saignement (niveaux liquide-liquide) (fig. 6-48). Sur le plan histopathologique, le lymphangiome est constitué de canaux vasculaires dilatés remplis de liquide clair ou de sang. Du fait du caractère diffus de la lésion, l’exérèse complète est en général impossible. Un traitement antibiotique et corticoïde des infections rhinopharyngées est parfois utile. Si l’exophtalmie est trop importante ou la vision menacée, un abord chirurgical peut être nécessaire en essayant de respecter les muscles oculomoteurs et le nerf optique. Les injections sclérosantes ne seraient pas sans danger dans l’orbite [11]. Plus récemment, l’utilisation de colle biologique (TISSEEL® fibrin glue) a été décrite pour limiter les récidives et les poussées évolutives [12], ainsi que pour les cas les plus réfractaires des injections de bléomycine [13].
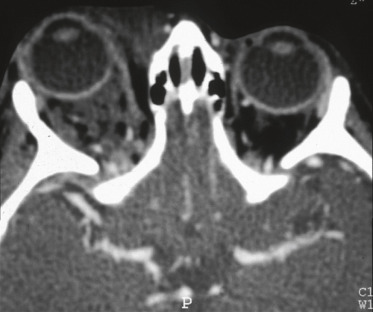
Fig. 6-48 Lymphangiome. Image de scanner.
Des kystes développés à partir de l’épithélium conjonctival peuvent parfois être rencontrés mais la lésion la plus fréquente est le kyste dermoïde. Ce sont des choristomes (malformation congénitale constituée de tissu normalement non présent dans l’organe). Ils sont découverts le plus souvent avant l’âge de 10 ans et ont tendance à se développer dans le quadrant supéro-externe de l’orbite ou au niveau du sourcil [14]. Ils se manifestent presque toujours sous forme d’une petite masse palpable. Lorsqu’ils sont situés au contact de l’os, ils peuvent provoquer une encoche osseuse. Les formes profondes représentent environ 30 % et sont plus difficiles à enlever chirurgicalement. Il est préférable de faire une imagerie avant d’envisager l’exérèse chirurgicale de la lésion afin de s’assurer de l’absence de lésion osseuse ou de prolongement postérieur du kyste dermoïde. L’exérèse doit être complète et en bloc, car la rupture peropératoire du kyste peut être responsable de réaction inflammatoire secondaire. Histologiquement, ils sont entourés d’un épithélium et contiennent des glandes sébacées, des follicules pileux, de la kératine et du cholestérol (fig. 6-49).
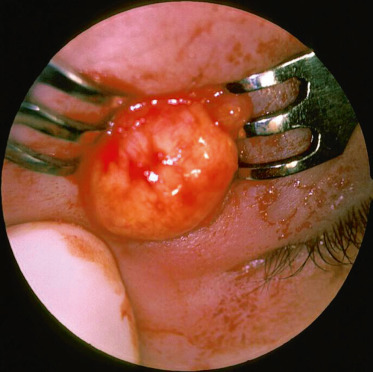
Fig. 6-49 Kyste dermoïde.
Le dermolipome n’est pas une lésion kystique mais c’est aussi un choristome pouvant toucher la conjonctive et parfois l’orbite. Cette malformation congénitale peut être associée au syndrome de Goldenhar. Les dermolipomes les plus volumineux doivent parfois être enlevés chirurgicalement, mais cette chirurgie devra être très prudente, car ils adhèrent fortement aux tissus normaux de l’orbite notamment aux muscles.
Les kystes colobomateux ne sont pas à proprement parler des tumeurs. Ce sont des malformations congénitales rares associant une microphtalmie avec un colobome. Le kyste peut être volumineux et peut parfois déformer la paupière. L’imagerie IRM permet de préciser la taille et l’étendue du kyste (qui apparaît sous la forme d’une masse arrondie) ainsi que ses rapports avec le globe malformé. Dans la plupart des cas cependant, surtout si le kyste est volumineux, l’exérèse chirurgicale du kyste et de l’oeil microphtalme est nécessaire avec mise en place d’un implant orbitaire ou d’une greffe dermograisseuse puis équipement prothétique.
Il s’agit d’une hernie des feuillets méningés, du liquide cérébrospinal et de cortex cérébral atrophique à travers une suture ou un orifice de l’orbite. Cette lésion congénitale peut provoquer une exophtalmie fluctuante augmentant lorsque l’enfant pleure. Le traitement est chirurgical.
C’est une tumeur d’origine germinale, congénitale et kystique très rare, qui peut se développer dans l’orbite. Elle entraîne dès la naissance une exophtalmie majeure, unilatérale, avec chémosis et déviation du globe oculaire. L’exophtalmie a souvent tendance à s’aggraver très vite. L’imagerie montre une masse multilobulée contenant typiquement de la graisse et des macrocalcifications s’étendant parfois à travers les parois orbitaires vers le sinus ou le cerveau. Histologiquement, la masse est constituée de kystes entourés d’épiderme, de muqueuse gastro-intestinale ou d’épithélium respiratoire.
Les neurofibromes orbitaires représentent l’une des lésions typiques de la maladie de Von Recklinghausen qui fait partie du groupe des phacomatoses. Il s’agit d’une maladie héréditaire transmise selon un mode autosomal dominant dont la fréquence serait de 1 sur 3 pour 4 000 naissances. Pour la neurofibromatose de type 1, le gène est localisé sur le bras long du chromosome 17. La neurofibromatose de type 2, qui intéresse essentiellement le système nerveux central, est due à une perte de gènes sur le chromosome 22. Elle associe des anomalies résultant d’une dystrophie neuro-ectodermale comprenant des troubles de la pigmentation cutanée et une variété de tumeurs provenant des cellules originaires de la crête neurale. Les atteintes orbitaires sont représentées par des neurofibromes, des gliomes du nerf optique et des méningiomes de la gaine du nerf optique. Des schwanomes peuvent se voir dans la neurofibromatose de type 2. Une dysplasie osseuse sphéno-orbitaire peut être associée. Les signes évocateurs de la maladie de Von Recklinghausen qui doivent être systématiquement recherchés lorsque le diagnostic est suspecté sont : à l’examen général, les nodules de Lish et la présence d’une tache café au lait ; en IRM, des îlots de dysplasie gliale sus- et sous-tentoriels caractéristiques.
Les localisations orbitaires de neurofibrome isolé sont associées à la maladie de Von Recklinghausen de type 1 dans environ 10 % des cas, mais presque 100 % des formes plexiformes sont associées à une maladie de Von Recklinghausen de type 2.
Le neurofibrome plexiforme est souvent présent avant l’âge de 10 ans. C’est une tumeur qui a la consistance d’un « amas de vermicelle » et qui a tendance à infiltrer les paupières et les tissus orbitaires. Un agrandissement de la cavité orbitaire visible sur les radiographies est souvent associé à un neurofibrome. Un envahissement des parties molles de l’hémiface peut se voir. Une IRM est indispensable pour préciser l’extension locorégionale de la tumeur qui va parfois jusqu’au sinus caverneux.
Le gliome du nerf optique et du chiasma est une tumeur rare associée dans environ 50 % des cas à une maladie de Von Recklinghausen de type 1 [15] et les gliomes bilatéraux sont plus fréquents chez ces patients. Histologiquement, ce sont le plus souvent des astrocytomes pilocytiques. Les signes cliniques sont une baisse de la vision associée à une exophtalmie axile en cas de gliome du nerf optique. Au FO, on observe d’abord un oedème papillaire puis une atrophie optique. Les gliomes de chiasma peuvent s’accompagner de troubles endocriniens. Les patients présentant une invasion précoce de l’hypothalamus avec un syndrome diencéphalique débutent généralement la maladie avant l’âge de 18 mois. Dans les gliomes du nerf optique, l’examen IRM permet en général de diagnostiquer une augmentation de volume fusiforme du nerf optique. Les indications thérapeutiques dépendent du degré de perte visuelle et d’exophtalmie, de la localisation de la tumeur et de son évolutivité. Une régression spontanée est possible. Actuellement, le traitement est le plus souvent conservateur.
Les méningiomes de la gaine du nerf optique ou du sphénoïde sont plus fréquents chez l’adulte mais des cas ont été décrits chez les enfants [16] et ils seraient plus fréquents en cas de maladie de Von Recklinghausen.
La dysplasie fibreuse de l’orbite est une maladie rare congénitale malformative qui est caractérisée par le remplacement du tissu osseux normal par un stroma fibrocellulaire contenant des îlots osseux immatures. Elle peut toucher plusieurs os dans l’organisme (forme polyostotique comme dans la maladie d’Albright caractérisée par une puberté précoce chez les filles et des taches pigmentées cutanées du côté atteint). Au niveau orbitaire, elle est souvent isolée (forme monostotique qui représente 80 % des cas). L’os frontal est le plus souvent atteint suivi par le sphénoïde et l’ethmoïde. L’épaississement de l’os frontal entraîne une exophtalmie, parfois inflammatoire avec déplacement du globe oculaire vers le bas et une asymétrie faciale. La maladie est lentement évolutive et peut avoir tendance à ralentir à la puberté.
Des kystes anévrismaux du toit de l’orbite et des fibromes ossifiant juvéniles des parois orbitaires peuvent se rencontrer et être responsables d’une exophtalmie. L’exérèse chirurgicale est le plus souvent nécessaire et nécessite une équipe pluridisciplinaire.
Les ostéosarcomes ne sont pas rares chez les enfants irradiés dans la petite enfance pour rétinoblastome bilatéral. La survenue de ces tumeurs est favorisée par l’irradiation mais également par la mutation du gène Rb1. On pense qu’environ 30 % des enfants porteurs du gène et irradiés développeront un sarcome radio-induit. Le pronostic vital de ces sarcomes est médiocre.
L’histiocytose à cellules de Langerhans ou histiocytose X est une maladie rare atteignant le plus souvent les nourrissons ou les enfants jeunes. L’étiologie n’est pas complètement élucidée mais impliquerait un dysfonctionnement immunologique. Les cellules de Langerhans sont une variante spécifique de l’histiocyte dendritique qui réside habituellement dans l’épiderme. La maladie peut être localisée ou comporter des atteintes multiples. Elle comporte dans 25 % des cas une atteinte orbitaire souvent sous forme de granulome éosinophile [17]. Le granulome éosinophile se présente comme un gonflement supérotemporal à début rapide, douloureux et inflammatoire (fig. 6-50). Le scanner montre un aspect typique avec une lésion ostéolytique au niveau de la paroi osseuse supérieure ou supérotemporale. La biopsie retrouve un aspect caractéristique avec une infiltration d’histiocytes de grande taille mononucléés et de cellules géantes de Touton. La positivité de la réaction à la protéine S100 en immunohistochimie, de même que la présence de granules de Birbeck en microscopie électronique permettent de différencier cette lésion du xanthogranulome juvénile. Un bilan général à la recherche d’atteintes systémiques doit être systématiquement réalisé. La numération-formule sanguine montre parfois une hyperéosinophilie. La maladie de Hand-Schuller- Christian est la forme chronique généralisée de l’histiocytose X. Elle est caractérisée par la triade classique diabète insipide, exophtalmie bilatérale et lacunes osseuses au niveau de la voûte crânienne. La maladie de Letterer-Siwe est une forme aiguë et multiviscérale d’histiocytose langerhancienne survenant chez le nourrisson entre 3 et 6 mois, rarement après 18 mois. Cette forme est de mauvais pronostic.
Les histiocytoses avec localisation orbitaire isolée ont un bon pronostic et guérissent généralement avec un curetage chirurgical. Les formes comportant une atteinte orbitaire plus sévère peuvent bénéficier d’une corticothérapie et/ou d’une radiothérapie avec une dose d’au plus 10 Gy. Enfin les formes avec atteintes systémiques sont parfois traitées par chimiothérapie.
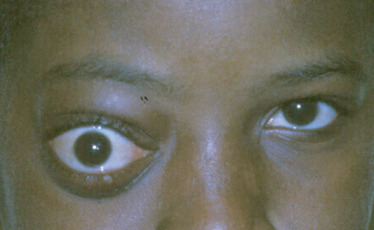
Fig. 6-50 Granulome éosinophile.
Le xanthogranulome juvénile est une pathologie bien connue des pédiatres produisant de multiples papules cutanées chez les nourrissons. Les manifestations ophtalmologiques comprennent une atteinte uvéale antérieure avec hyphéma et glaucome secondaire et des localisations orbitaires. Histologiquement, il existe une prolifération anormale des histiocytes avec des lymphocytes, des éosinophiles et surtout la présence caractéristique de cellules géantes de Touton. Les atteintes orbitaires sont rares. Elles surviennent le plus souvent chez un nourrisson et se manifestent par une exophtalmie uni- ou bilatérale. En l’absence de manifestation cutanée ou irienne caractéristique, une biopsie peut être nécessaire au diagnostic. Si l’exérèse chirurgicale complète de la lésion orbitaire est possible, celle-ci doit être privilégiée. Si l’exérèse est incomplète, un traitement corticoïde postopératoire est en général nécessaire.
Ces atteintes représentent une cause non exceptionnelle d’exophtalmie. Elles sont plus fréquentes avec les formes aiguës qu’avec les formes chroniques et avec les formes lymphoblastiques qu’avec les formes myéloblastiques. Les signes cliniques comportent une exophtalmie, un oedème des paupières, un chémosis et des douleurs. L’atteinte peut être uni- ou bilatérale. Elle peut être en rapport avec la présence d’une masse constituée de cellules leucémiques ou avec une hémorragie orbitaire. L’atteinte orbitaire est souvent présente à la phase terminale de la maladie mais peut parfois être le signe révélateur. Dans les leucémies myéloblastiques, la masse orbitaire correspond à un sarcome granulocytaire ou chlorome.
Il s’agit d’une affection associée à la présence du virus Epstein-Barr plus fréquent en Afrique tropicale. Dans 60 % des cas, il existe une tumeur du maxillaire envahissant l’orbite et causant une exophtalmie massive. Les patients présentant une atteinte abdominale ont un pronostic plus péjoratif, mais la survie globale a été améliorée grâce à l’utilisation d’une polychimiothérapie.
La cause la plus fréquente de métastase orbitaire chez l’enfant est le neuroblastome. La tumeur primitive est le plus souvent diagnostiquée avant la survenue de la métastase orbitaire mais dans 10 % des cas, la métastase est révélatrice. L’atteinte métastatique siège sur les structures osseuses autour des sutures, mais s’étend par contiguïté aux tissus mous en refoulant l’ensemble du contenu orbitaire (syndrome de Hutchinson). L’exophtalmie peut être bilatérale avec ecchymose périorbitaire. Le neuroblastome est une tumeur maligne du système nerveux sympathique. La tumeur primitive peut siéger au niveau des surrénales ou d’autres structures rétropéritonéales mais également les chaînes cervicales, le médiastin, le pelvis. Quatre-vingt-dix pour cent des enfants ont un taux élevé de catécholamines urinaires. Le traitement repose sur la polychimiothérapie [18].
Le sarcome d’Ewing est une tumeur rare pouvant également être à l’origine de métastase orbitaire. C’est la deuxième tumeur maligne osseuse en fréquence chez l’enfant. Des formes primitives de l’orbite ont été rapportées. Dans cette tumeur, les études biologiques ont montré une surexpression de CD99 et d’une protéine EWS-FLI1. Ce sont des biomarqueurs importants qui pourraient représenter dans l’avenir des cibles thérapeutiques.
Le rétinoblastome est une tumeur intra-oculaire qui peut se propager à l’orbite, soit en raison d’une absence de traitement si la tumeur n’est pas diagnostiquée à temps, soit du fait d’une récidive orbitaire après énucléation. Pour éviter ces récidives orbitaires, il faut faire pratiquer un examen anatomopathologique soigneux du globe énucléé et compléter le traitement par chimiothérapie et/ou radiothérapie en cas d’extension au nerf optique ou d’extension extrasclérale. Dans les pays en voie de développement, les rétinoblastomes avec extension à l’orbite sont fréquents et constituent une forme de présentation courante de la maladie.
Le médullo-épithéliome du corps ciliaire peut aussi se présenter sous une forme agressive capable de récidiver dans l’orbite et de métastaser.
Le mélanome uvéal est exceptionnel chez l’enfant de moins de 16 ans. L’envahissement orbitaire peut se voir dans les formes évoluées qui nécessitent alors une énucléation élargie suivie d’irradiation orbitaire.
Le diagnostic des tumeurs orbitaires de l’enfant repose sur un interrogatoire et un examen clinique soigneux complété au besoin par un bilan général (numération-formule sanguine, radiographie du thorax échographie abdominale). Actuellement, les examens radiologiques, et principalement l’IRM, permettent une localisation exacte de la lésion et un bilan d’extension locorégionale précis. Dans certains cas, il n’est pas nécessaire de faire une biopsie orbitaire (tableau d’hémangiome capillaire bénin, découverte d’une masse abdominale orientant vers une métastase de neuroblastome). Ailleurs, comme dans le rhabdomyosarcome, la biopsie doit être faite en urgence pour confirmer le diagnostic et mettre en route la chimiothérapie. Le concours d’anatomopathologistes habitués aux tumeurs de l’enfant est souvent utile.
Remerciements : Christine Levy-Gabriel.
[1] Kodsi SR, Shetlar DJ, Campbell RJ, et al. A review of 340 orbital tumors in children during a 60-year period. Am J Ophthalmol 1994 ; 117 : 177-82.
[2] Shields JA, Shields CL, Scartozzi R. Survey of 1264 patients with orbital tumors and simulating lesions : The 2002 Montgomery Lecture, part 1. Ophthalmology 2004 ; 111 : 997-1008.
[3] Klijanienko J, Couturier J, Bourdeaut F, et al. Fine-needle aspiration as a diagnostic technique in 50 cases of primary Ewing sarcoma/peripheral neuroectodermal tumor. Institut Curie’s experience. Diagn Cytopathol 2012 ; 40 : 19-25.
[4] Merks JH, De Salvo GL, Bergeron C, et al. Parameningeal rhabdomyosarcoma in pediatric age : results of a pooled analysis from North American and European cooperative groups. Ann Oncol 2014 ; 25 : 231-6.
[5] Shern JF, Chen L, Chmielecki J, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusionpositive and fusion-negative tumors. Cancer Discov 2014 ; 4 : 216-31.
[6] Oberlin O, Rey A, Sanchez de Toledo J, et al. Randomized comparison of intensified six-drug versus standard three-drug chemotherapy for high-risk nonmetastatic rhabdomyosarcoma and other chemotherapy-sensitive childhood soft tissue sarcomas : long-term results from the International Society of Pediatric Oncology MMT95 study. J Clin Oncol 2012 ; 30 : 2457-65.
[7] Boutroux H, Levy C, Mosseri V, et al. Long-term evaluation of orbital rhabdomyosarcoma in children. Clin Experiment Ophthalmol 2015 ; 43 : 12-9.
[8] Schwartz SR, Blei F, Ceisler E, et al. Risk factors for amblyopia in children with capillary hemangiomas of the eyelids and orbit. J AAPOS 2006 ; 10 : 262-8.
[9] Burne R, Taylor R. Monitoring propranolol treatment in periocular infantile haemangioma. Eye (Lond) 2014 ; 28 : 1281-4 ; quiz 1285.
[10] Bailey ST, Wojno TH, Shields CL, Grossniklaus HE. Late-onset presentation of orbital lymphangioma. Ophthal Plast Reconstr Surg 2007 ; 23 : 100-3.
[11] Schwarcz RM, Ben Simon GJ, Cook T, Goldberg RA. Sclerosing therapy as first line treatment for low flow vascular lesions of the orbit. Am J Ophthalmol 2006 ; 141 : 333-9.
[12] Boulos PR, Harissi-Dagher M, Kavalec C, et al. Intralesional injection of Tisseel fibrin glue for resection of lymphangiomas and other thin-walled orbital cysts. Ophthal Plast Reconstr Surg 2005 ; 21 : 171-6.
[13] Gooding C, Meyer D. Intralesional bleomycin : a potential treatment for refractory orbital lymphangiomas. Ophthal Plast Reconstr Surg 2014 ; 30 : e65-7.
[14] Cavazza S, Laffi GL, Lodi L, et al. Orbital dermoid cyst of childhood: clinical pathologic findings, classification and management. Int Ophthalmol 2011 ; 31 : 93-7.
[15] Thiagalingam S, Flaherty M, Billson F, North K. Neurofibromatosis type 1 and optic pathway gliomas : follow-up of 54 patients. Ophthalmology 2004 ; 111 : 568-77.
[16] Amirjamshidi A, Mehrazin M, Abbassioun K. Meningiomas of the central nervous system occurring below the age of 17 : report of 24 cases not associated with neurofibromatosis and review of literature. Childs Nerv Syst 2000 ; 16 : 406-16.
[17] McCaffrey J, Rawlingson C. Multisystem langerhans cell histiocytosis with advanced orbital involvement : case report. East Afr Med J 2010 ; 87 : 430-2.
[18] Smith SJ, Diehl NN, Smith BD, Mohney BG. Incidence, ocular manifestations, and survival in children with neuroblastoma : a population-based study. Am J Ophthalmol 2010 ; 149 : 677-82.e2.
M. Callet, A. Gallucci , O. Galatoire, F. Cheynet, D. Denis
Les infections orbitaires de l’enfant sont rares mais potentiellement très graves pouvant même, lorsqu’elles s’étendent en arrière de l’orbite, être responsables de complications intracrâniennes.
Leur prise en charge est urgente et doit être globale, impliquant une coopération entre ophtalmologistes, pédiatres, chirurgiens maxillofaciaux, oto-rhino-laryngologistes et radiologues.
Le diagnostic de la forme clinique et d’une étiologie parfois évidente est essentiel dans une bonne prise en charge de l’enfant. Le lecteur pourra également se référer au chapitre 27-13.
La pathologie infectieuse orbitaire témoigne des rapports étroits qui unissent l’oeil et l’orbite. L’orbite est en contact avec les cavités sinusiennes par ses faces internes, supéro-internes et inférieures. Son sommet est partiellement constitué par le sinus sphénoïdal et les cellules ethmoïdales postérieures. Les parois osseuses sont minces et fragiles, et il y a des communications canaliculaires directes par les vaisseaux et les nerfs perforants, ce qui explique la facilité de passage des processus infectieux (à la différence des artères et des lymphatiques, les veines ont une importance considérable pour le passage des processus pathologiques). De nombreuses veinules existent entre sinus et orbite, dont les plus importantes viennent de la région ethmoïdale et frontale, pour se jeter dans la veine ophtalmique supérieure, et de la région maxillaire, pour se jeter dans la veine ophtalmique inférieure.
Les cellules ethmoïdales apparaissent chez le foetus de 4 mois et continuent progressivement leur développement pour être formées vers 7 ans et se terminer vers 12-14 ans.
Le sinus maxillaire, présent à la naissance se développe jusqu’à 7 ans.
Le sinus frontal après être apparu entre 5 et 7 ans a son volume définitif vers 25 ans.
Le sinus sphénoïdal se développe entre 4 et 5 ans (fig. 6-51 et fig. 6-52). Les cellules ethmoïdales étant les premières à apparaître, les pathologies infectieuses les plus fréquentes dans les premières années de vie sont d’origine ethmoïdale. Les atteintes du maxillaire surviennent un peu plus tardivement et celles du sinus frontal encore plus tardivement, vers 9-10 ans. Enfin, les atteintes du sinus sphénoïdal sont plus rares et rarement isolées.
La cellulite orbitaire représente 0,9 cas pour 1 000 admissions en pédiatrie. L’âge moyen de survenue est situé entre 6 et 7 ans, avec une prépondérance masculine [1, 2].
Elle implique une infection de l’espace pré- ou rétroseptal et résulte généralement d’une infection de voisinage. La porte d’entrée des cellulites orbitaires est le plus souvent sinusienne (85 % ), surtout ethmoïdale entre 6 mois et 5 ans, maxillaire à partir de 3 ans, et frontale après l’âge de 10 ans. Elle peut être aussi cutanée (10 % ) mais toutes les causes d’infections locorégionales peuvent être retrouvées (5 % ) : il faut se méfier d’une présentation atypique causée par un traumatisme orbitaire pénétrant avec rétention de corps étranger [3], une pathologie lacrymale telle un dacryocystocèle ou une dacryocystite extensive [4], d’un abcès dentaire [5], de toute chirurgie oculo-orbitaire [6] et également garder en tête la possibilité d’une tumeur orbitaire révélée à cette occasion tel un rhabdomyosarcome [4].
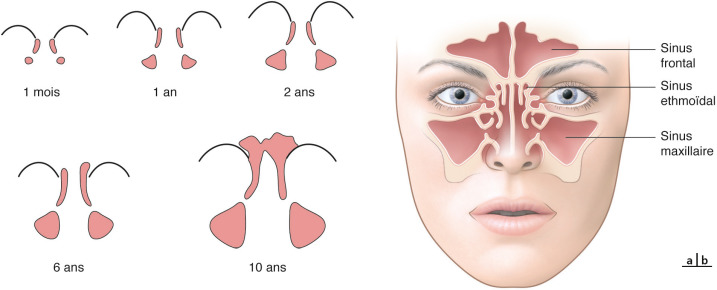
Fig. 6-51 Développement des sinus avec cavitation progressive selon l’âge.
a. Anatomie du sinus de l’enfant. b. Anatomie du sinus de l’adulte.
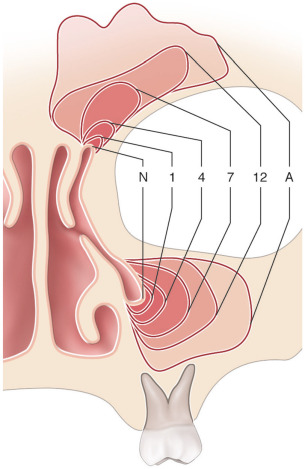
Fig. 6-52 Formation des sinus en fonction de l’âge, de la naissance (N) à l’âge adulte (A) (âge exprimé en années) d’après Arey.
Son diagnostic est clinique avec mise en évidence d’un syndrome orbitaire chez un enfant associant un oedème inflammatoire des paupières, qui sont tendues nécessitant parfois une manipulation de la région pour examiner le globe oculaire, une rougeur palpébrale, avec parfois un chémosis, le tout évoluant rapidement dans un contexte de sinusite, plus précisément d’éthmoïdite (98 % ) [7] ou de pansinusite.
Les signes ophtalmologiques de gravité sont à rechercher : ophtalmoplégie douloureuse partielle ou totale, ptosis, exophtalmie, neuropathie optique.
L’examen des voies lacrymales recherche une dacryocystite et une inspection soigneuse de la région du sac lacrymal permet de poser le diagnostic différentiel étiologique entre une dacryocystite et une sinusite maxillaire ; celle-ci entraîne une tuméfaction plus marquée de la paupière inférieure.
L’examen maxillofacial recherche une affection des sinus ou d’origine dentaire : il comprend une rhinoscopie antérieure, un examen du palais, des gencives ou encore la palpation des sinus, et recherche une rhinorrhée purulente unilatérale, la présence de collection purulente au niveau du méat moyen qui sont fortement évocateurs.
Suivant la localisation de la tuméfaction au début de l’affection, il est parfois possible de localiser l’origine probable de la sinusite :
une origine ethmoïdale se manifeste dans la région de l’angle interne, au-dessus du ligament latéral (fig. 6-53) ;
une origine frontale a une localisation très proche, éventuellement un peu plus haute, sauf si le processus touche le récessus temporal, ce qui entraîne une tuméfaction de la partie externe de la paupière supérieure.
L’examen pédiatrique est indispensable dans ce contexte infectieux. Il apprécie l’état général de l’enfant (fièvre, comportement) et recherche des signes neurologiques de gravité (pour exemple, infection à Haemophilus).
L’objectif de ces examens est de prévenir rapidement une détérioration rapide et des séquelles sévères telles que la baisse de vision, la thrombose du sinus caverneux, l’abcès cérébral, l’ostéomyélite et la septicémie.
Fig. 6-53 a, b. Cellulite orbitaire compliquant une ethmoïdite aiguë infectieuse droite chez un enfant.
Les germes retrouvés sont habituellement ceux pourvoyeurs de sinusites de l’enfant : Haemophilus influenzae type B dont la fréquence a considérablement diminué depuis l’avènement du vaccin mais qui persiste à être pourvoyeur de sinusites compliquées [8, 9], et les cocci à Gram positif tels Staphylococcus aureus et Epidermidis, Streptococcus Pneumoniae, Streptococcus pyogenes, Moraxella catarrhalis [10], sans oublier Staphylococcus aureus résistant à la méthicilline [11]. Les infections polymicrobiennes et à germes anaérobies sont plus fréquentes chez les enfants plus âgés.
L’imagerie est indispensable en cas de suspicion de cellulite orbitaire.
La radiographie standard n’est plus indiquée, bien qu’elle montre le foyer de sinusite.
Une TDM orbitaire avec et sans injection de produit de contraste est l’examen clé du diagnostic positif et parfois étiologique, mais ne permet pas toujours de différencier abcès, oedème inflammatoire et hématome. En revanche, la TDM permettra de déterminer la localisation exacte, la taille de la lésion orbitaire et l’état des sinus de la face (fig. 6-54).
L’IRM orbitaire est indiquée dans les cas complexes, lors de forte suspicion clinique sans imagerie évocatrice au scanner ou en cas de complications intracrâniennes suspectées, telles qu’un abcès cérébral ou une méningite. L’IRM avec injection de gadolinium objective, typiquement dans les cellulites orbitaires, une infiltration inflammatoire se traduisant par un aspect hypo-intense en T1, hyper-intense en T2. En séquence T1 avec injection de gadolinium, il existe un rehaussement intra- et périorbitaire. Un abcès sous-périosté est décrit comme une lésion iso-intense avec les zones nécrotiques hypo-intenses en T1 et une hyper-intensité en T2 sans gadolinium. Après injection de gadolinium en T1, la lésion est rehaussée en périphérie, avec maintien d’un hyposignal dans les zones centrales nécrotiques. L’abcès orbitaire est décrit à l’IRM comme un abcès nécrotique avec une hypo-intensité T1 et hyper-intensité T2.
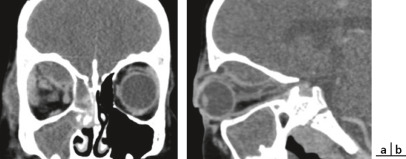
Fig. 6-54 TDM orbitaire d’une cellulite orbitaire droite compliquant une ethmoïdite chez un enfant de 4 ans : vues de face (a) et de profil (b).
(encadrés 6-3 et 6-4)
Une classification en cinq stades, fondée sur l’anatomie, a été proposée par Chandler [12], toutes pouvant s’associer, et ne progressant pas forcément par ordre : cellulite préseptale (I), cellulite orbitaire (II), abcès sous-périosté (III), abcès orbitaire (IV), thrombose du sinus caverneux (V) (Tableau 6-8 et fig. 6-55).
La conduite à tenir dans une suspicion de cellulite orbitaire est proposée à la fig. 6-56.
Cette classification est aussi une gradation de l’évolution de la cellulite. Ainsi, une cellulite préseptale (grade I) peut évoluer en l’absence de traitement vers un grade IV voire V. Cependant, cette évolution graduelle n’est pas systématique.
Suivant la physiopathogénie, il est également possible de distinguer les cellulites en :
préseptales (grade I) des cellulites orbitaires vraies ;
rétroseptales (grades II à V).
Examen d’un enfant avec suspicion de cellulite orbitaire
Sexe, âge.
Signes de gravité : ophtalmologiques, locorégionaux, généraux.
Étiologie, porte d’entrée : sinusite, infection des voies lacrymales, conjonctivite, tumeur, traumatisme.
Examen ophtalmologique :
-
préseptale ou rétroseptale ;
latéralité ;
acuité visuelle initiale ;
oedème palpébral ;
exophtalmie, ophtalmoplégie ;
oedème papillaire.
Examen ORL : rhinoscopie, rhinorrhée, otalgie.
Examen maxillofacial : oedème palpébral, collection, abcès, exophtalmie.
Examen général pédiatrique : notion de fièvre, état général.
Paraclinique :
-
biologie : leucocytose, CRP, bactériologie ;
imagerie : TDM ± IRM selon contexte.
Étiologies des cellulites orbitaires de l’enfant
Sinusites +++ : ethmoïdite ou pansinusite.
Infections des voies lacrymales : dacryocystite, dacryocystocèle congénital.
Conjonctivites :
-
bactériennes (24 % ) ;
virales (16 % ) ;
germes atypiques.
Tumeurs : rhabdomyosarcome, rétinoblastome.
Traumatisme.
Fig. 6-55 Classification de Chandler, d’après EMC et Mouriaux et al. [2].
a. Grade I : cellulite préseptale ou oedème inflammatoire orbitaire. b. Grade II : cellulite orbitaire diffuse. c. Grade III : abcès sous-périosté. d. Grade IV : abcès orbitaire. (Grade V non illustré : thrombose du sinus caverneux.)
Tableau 6-8 – Classifi cation des cellulites orbitaires d’après Chandler.
C’est la forme clinique la plus fréquente (72 à 96 % des cas) [13] et la moins grave, car l’infection est circonscrite en avant du septum orbitaire. Souvent unilatérale, elle survient surtout chez les enfants de moins de 5 ans. L’oedème palpébral est dû à l’obstruction du plexus de la veine ophtalmique supérieure [2]. Elle est souvent secondaire à une infection cutanée de la paupière (orgelet, impétigo, érysipèle, herpès, varicelle, plus rarement à une dacryocystite) mais aussi à une infection des voies respiratoires hautes, une sinusite non compliquée, une otite moyenne aiguë ou un traumatisme palpébral.
Elle doit être distinguée des autres causes d’oedème palpébral telles que :
la conjonctivite à adénovirus [14] : il a été rapporté jusqu’à 16 % d’erreur diagnostique chez des enfants étiquetés comme présentant une cellulite préseptale, étant en réalité des conjonctivites à adénovirus ;
la conjonctivite atopique ;
le syndrome de Kawasaki [15], plus rare.
Il existe, dans 30 % des cas, une exophtalmie modérée due à un oedème inflammatoire de l’orbite. La douleur est peu intense et est décrite plutôt comme une tension des tissus sous-cutanés [2]. À l’examen, l’oeil est blanc, pouvant présenter un chémosis modéré dans de rares cas et le FO normal. L’oedème palpébral est marqué, purulent ou non selon le germe, pouvant s’étendre à la joue ou au sourcil. L’acuité visuelle est conservée, il n’existe pas d’exophtalmie, ni de limitation de l’oculomotricité et la pupille est normale.
Parmi les agents pathogènes, une particularité doit être soulignée un tableau infectieux systémique souvent marqué, avec possibilité d’une bactériémie associée voire une méningite. On retrouve un tableau de cellulite préseptale non suppurative, avec une coloration bleuâtre ou violette de la paupière.pour l’infection à Haemophilus influenzae : l’enfant présente
Sur le plan thérapeutique, en cas de cellulite préseptale non collectée, la prise en charge peut être faite en externe avec une antibiothérapie orale. L’évolution clinique guide la prise en charge secondaire : une amélioration clinique conduit au prolongement du traitement per os au domicile pendant au minimum 10 jours. Une évolution défavorable impose une prise en charge similaire à celle d’une cellulite rétroseptale, notamment une antibiothérapie par voie intraveineuse. En cas de cellulite préseptale collectée, l’incision de l’abcès sous anesthésie générale est de règle avec, si possible, la mise en place d’un drainage.
Toujours grave et potentiellement menaçante pour le pronostic vital et visuel, la cellulite orbitaire diffuse est plus fréquente chez les enfants de plus de 5 ans (âge moyen 7 ans), en période hivernale. Les signes cliniques sont liés à la fois à l’inflammation intra-orbitaire et à une infiltration bactérienne, parfois difficile à mettre en évidence. La douleur est profonde. L’examen retrouve là encore un oedème palpébral important, qui est diffus au niveau de l’orbite par infiltration des micro-organismes et des cellules inflammatoires et la sensibilité cutanée peut être diminuée.
Fig. 6-56 Conduite à tenir devant une suspicion de cellulite orbitaire pédiatrique.
Les signes de dysfonction orbitaire sont : exophtalmie, chémosis, hyperhémie conjonctivale, ptosis, limitation douloureuse des mouvements oculaires, neuropathie optique.
Les complications qu’elle peut entraîner sont : neuropathie optique, atrophie optique, kératite d’exposition, occlusion de l’artère centrale de la rétine [16], ischémie rétinienne et choroïdienne, abcès sous-périosté et orbitaire [17], thrombose du sinus caverneux, méningite, abcès cérébral et septicémie.
Sur le plan oculomoteur, l’exophtalmie est axile, la limitation de l’oculomotricité est due à l’oedème de l’orbite et/ou à une atteinte musculaire toxique due aux micro-organismes. Une atteinte des nerfs crâniens III, IV, VI est possible, et ce d’autant qu’il existe une atteinte de la fissure orbitaire supérieure et du sinus caverneux.
Sur le plan sensoriel, une baisse d’acuité visuelle est possible. Elle peut être attribuée à : la neuropathie optique, une kératite d’exposition qu’il faut rechercher ou une occlusion vasculaire.
Sur le plan thérapeutique, elle nécessite une prise en charge urgente avec administration intraveineuse d’une bi-antibiothérapie à forte dose (généralement céphalosporine de troisième génération combinée avec pénicilline-acide clavulanique, métronidazole, clindamycine). Elle ne sera retardée ni par les prélèvements microbiologiques, ni par la TDM cérébro-orbitaire, qui précisera l’extension à l’orbite, au périoste et au parenchyme cérébral de la pathologie, et un éventuel point de départ sinusien.
L’introduction de la corticothérapie semble envisageable à partir d’un certain seuil de C-reactive proteine (CRP) établi à moins de 4 mg/dL et permettrait une amélioration plus rapide de la symptomatologie [18].
L’indication d’un drainage chirurgical sera guidé par la clinique et par les stades de Chandler, avec la notion récente que la présence d’une lyse osseuse et des dimensions de l’abcès sous-périosté objectivés en TDM orienteraient la prise en charge chirurgicale [19].
Il faut suspecter un abcès lorsqu’il existe un sepsis marqué, une réaction orbitaire majeure ou devant une réponse insuffisante à une antibiothérapie intraveineuse bien conduite. On retrouve majoritairement des germes aérobies, bien que certains germes anaérobies puissent être responsables [20]. L’abcès résulte d’une accumulation de débris inflammatoires et de bactéries sous le périoste orbitaire, témoin de l’envahissement de l’orbite à partir d’un foyer de sinusite adjacent, avec un oedème localisé autour de cet abcès. Dans les abcès sous-périostés, l’infection purulente d’un sinus (ethmoïdal le plus souvent) fait irruption à travers la lame papyracée sous le périoste, qui est facilement soulevé de l’os [21] donnant une apparence convexe « en lentille » en TDM. La douleur orbitaire n’est pas constante. La palpation de l’orbite peut retrouver une douleur exquise et une masse si l’abcès est antérieur ou volumineux. Les signes compressifs sont variables en fonction de la localisation de l’abcès : exophtalmie non axile avec un déplacement opposé à l’abcès, baisse d’acuité visuelle en cas de compression du nerf optique, troubles oculomoteurs.
Sur le plan thérapeutique, les abcès sous-périostés peuvent être traités par antibiothérapie seule, surtout chez les jeunes patients, si l’épaisseur de la collection est inférieure à 10 mm sans effet de masse sur le droit interne et en l’absence de bulles d’air évoquant une infection anaérobie []. Les taux de réussite sont estimés à 93 % [23].
Dans tous les autres cas, notamment en présence de signes ophtalmologiques comme une baisse d’acuité visuelle, un déficit pupillaire afférent (signant une neuropathie optique), des troubles oculomoteurs importants, un patient âgé, un abcès collecté intra-orbitaire ou une aggravation du tableau clinique, la chirurgie est prônée. La chirurgie repose habituellement sur la réalisation d’une orbitotomie par voie antérieure, puis éventuellement après ponction de l’abcès, l’ouverture-drainage de la collection. Une lame de Silastic® ou des crins de Florence peuvent être placés localement pendant 24 à 48 heures. L’évacuation de l’abcès peut aussi se faire par voie endonasale (avec une équipe entraînée), après avoir effondré la lame papyracée [24].
Un abcès orbitaire résulte d’une brèche infectieuse dans le périoste ou d’un ensemencement intra-orbitaire. La douleur est profonde, l’oedème palpébral est majeur et l’exophtalmie constante. Celle-ci peut être axile ou non en fonction de la localisation de l’abcès. La sensibilité cutanée est diminuée. Les troubles oculomoteurs peuvent aller jusqu’à l’ophtalmoplégie complète et il existe de façon très fréquente une baisse d’acuité visuelle.
Sur le plan ophtalmologique, l’examen retrouve un chémosis important, une pupille en mydriase. L’examen du fond d’oeil retrouve parfois un oedème papillaire avec dilatation veineuse ou des plis choroïdiens. L’extension de l’infection au système nerveux peut provoquer une méningite ou un abcès cérébral.
Sur le plan thérapeutique, la localisation de l’abcès guide l’approche chirurgicale, rendant possible un drainage endoscopique endonasal ou une voie externe [25].
La thrombose du sinus caverneux est la complication majeure à redouter.
La thrombose du sinus caverneux peut être uni- ou bilatérale : elle témoigne de l’extension postérieure de l’infection via les veines ophtalmiques supérieures.
Sur le plan général, la douleur est profonde. On note une fièvre et des céphalées dans deux tiers des cas. Il peut exister une atteinte méningée avec sepsis et altération de la conscience : le pronostic vital est alors mis en jeu.
Sur le plan ophtalmologique, l’oedème est majeur avec chémosis, ophtalmoplégie avec atteinte V1 et V2, voire neuropathie. Les signes de congestion des paupières sont présents, qui prennent un aspect bleu violacé. L’exophtalmie est majeure et l’acuité visuelle est effondrée. L’atteinte nerveuse est globale, avec une pupille en mydriase, une ophtalmoplégie complète (atteinte du III, du IV, du VI). L’examen du fond d’oeil révèle un oedème papillaire ou des signes d’occlusion veineuse par compression extrinsèque.
Ces signes cliniques ne sont pas spécifiques de l’infection : une thrombophlébite peut survenir dans un contexte d’hypercoagulabilité ou de traumatisme, notamment chez des jeunes adultes [26, 27].
Les symptômes sont comparables à ceux d’une thrombose du sinus caverneux, mais il s’agit d’une ophtalmoplégie douloureuse récidivante, parfois alternante.
L’examen anatomopathologique réalisé sur biopsie montre un tissu inflammatoire granulomateux non spécifique, infiltrant le sinus caverneux. En l’absence de biopsie, le syndrome de Tolosa- Hunt est un diagnostic d’élimination, dont le traitement repose sur la corticothérapie [28].
[1] Huang SF, Lee TJ, Lee YS, et al. Acute rhinosinusitis-related orbital infection in pediatric patients : a retrospective analysis. Ann Otol Rhinol Laryngol 2011 ; 120 : 185-90.
[2] Mouriaux F, Rysaneka B, Babin E, Cattoir V. Les cellulites orbitaires. J Fr Ophtalmol 2012 ; 35 : 52-7.
[3] Gawdat TI, Ahmed RA. Orbital foreign bodies : expect the unexpected. J Pediatr Ophthalmol Strabismus 2010 ; 47 Online : e1-4.
[4] Promelle V, Bennai D, Drimbea A, et al. Cellulites orbitaires atypiques d’origine non sinusienne de l’enfant : à propos de quatre cas. J Fr Ophtalmol 2014 ; 37 : 149-54.
[5] De Assis-Costa MD, Santos GS, Maciel J, et al. Odontogenic infection causing orbital cellulitis in a pediatric patient. J Craniofac Surg 2013 ; 24 : 526-9.
[6] Mikhail M, Koenekoop RK, Khan A. Orbital cellulitis and multiple abscess formation after strabismus surgery. Can J Ophthalmol 2016 ; 51 : e60-2.
[7] Boivin L, Adenis JP. Infections orbitaires de l’enfant : clinique, imagerie et traitement. J Fr Ophtalmol 2009 ; 32 : 368-73.
[8] Sharma A, Liu ES, Le TD, et al. Pediatric orbital cellulitis in the Haemophilus influenzae vaccine era. J AAPOS 2015 ; 19 : 206-10.
[9] Rimon A, Hoffer V, Prais D, et al. Periorbital cellulitis in the era of Haemophilus influenzae type B vaccine : predisposing factors and etiologic agents in hospitalized children. J Pediatr Ophthalmol Strabismus 2008 ; 45 : 300-4.
[10] McKinley SH, Yen MT, Miller AM, Yen KG. Microbiology of pediatric orbital cellulitis. Am J Ophthalmol 2007 ; 144 : 497-501.
[11] Weiss A, Friendly D, Eglin K, et al. Bacterial periorbital and orbital cellulitis in childhood. Ophthalmology 1983 ; 90 : 195-203.
[12] Chandler JR, Langenbrunner DJ, Stevens ER. The pathogenesis of orbital complications in acute sinusitis. Laryngoscope 1970 ; 80 : 1414-28.
[13] Sobol SE, Marchand J, Tewfik TL, et al. Orbital complications of sinusitis in children. Pediatr Infect Dis J 2006 ; 25 : 695-9.
[14] Ruttum MS, Ogawa G. Adenovirus conjunctivitis mimics preseptal and orbital cellulitis in young children. Pediatr Infect Dis J 1996 ; 15 : 266-7.
[15] Sheard RM, Pandey KR, Barnes ND, Vivian AJ. Kawasaki disease presenting as orbital cellulitis. J Pediatr Ophthalmol Strabismus 2000 ; 37 : 123-5.
[16] Proctor CM, Magrath GN, de Castro LE, et al. Orbital cellulitis complicated by central retinal artery occlusion. Ophthal Plast Reconstr Surg 2013 ; 29 : e59-61.
[17] Jain A, Rubin PA. Orbital cellulitis in children. Int Ophthalmol Clin 2001 ; 41 : 71-86.
[18] Davies BW, Smith JM, Hink EM, Durairaj VD. C-Reactive Protein as a marker for initiating steroid treatment in children with orbital cellulitis. Ophthal Plast Reconstr Surg 2015 ; 31 : 364-8.
[19] Le TD, Liu ES, Adatia FA, et al. The effect of adding orbital computed tomography findings to the Chandler criteria for classifying pediatric orbital cellulitis in predicting which patients will require surgical intervention. J AAPOS 2014 ; 18 : 271-7.
[20] Brook I. Microbiology and choice of antimicrobial therapy for acute sinusitis complicated by subperiosteal abscess in children. Int J Pediatr Otorhinolaryngol 2016 ; 84 : 21-6.
[21] Eviatar E, Sandbank J, Kleid S, Gavriel H. The role of osteitis of the lamina papyracea in the formation of subperiosteal orbital abscess in young children. Int J Pediatr Otorhinolaryngol 2014 ; 78 : 2267-70.
[22] Ryan JT, Preciado DA, Bauman N, et al. Management of pediatric orbital cellulitis in patients with radiographic findings of subperiosteal abscess. Otolaryngol Head Neck Surg 2009 ; 140 : 907-11.
[23] Catalano RA, Smoot CN. Subperiosteal orbital masses in children with orbital cellulitis : time for a reevaluation ? J Pediatr Ophthalmol Strabismus 1990 ; 27 : 141-2.
[24] Bhargava D, Sankhla D, Ganesan A, Chand P. Endoscopic sinus surgery for orbital subperiosteal abscess secondary to sinusitis. Rhinology 2001 ; 39 : 151-5.
[25] Gavriel H, Jabrin B, Eviatar E. Management of superior subperiosteal orbital abscess. Eur Arch Otorhinolaryngol 2016 ; 273 : 145-50.
[26] Frank GS, Smith JM, Davies BW, et al. Ophthalmic manifestations and outcomes after cavernous sinus thrombosis in children. J AAPOS 2015 ; 19 : 358-62.
[27] Oxford Le, McClay J. Complications of acute sinusitis in children. Otolaryngol Head Neck Surg 2005 ; 133 : 32-7.
[28] Orssaud C, Roche O, El Dirani H, et al. Painful ophtalmoplegia in children : Tolosa Hunt syndrome or ophtalmologic migraine ? Arch Pediatr 2007 ; 14 : 996-9.