Principales pathologies systémiques responsables de troubles ophtalmologiques
Coordonné par M. Robert, C. Speeg-Schatz
C. Orssaud, M. Robert
Les anomalies chromosomiques sont favorisées par l'âge avancé de la mère, contrairement aux néomutations génétiques liées à l'âge avancé du père.
La trisomie 13, ou syndrome de Bartholin-Patau, est caractérisée par l'association d'une microcéphalie avec holoprosencéphalie responsable d'un déficit intellectuel sévère, d'une dysmorphie faciale, d'une polydactylie postaxiale, d'une cardiopathie et de malformations oculaires. Environ 90 % des enfants nés vivants décèdent avant l'âge de 1 an. À l'extrême, la dysmorphie faciale réalise une cébocéphalie, avec un œil unique et malformé au bout d'un proboscis (trompe) à la place du nez. Le syndrome est connu des ophtalmologistes pour être la cause principale de la dysplasie rétinienne de Reese, qui constitue une forme particulière de persistance de la vascularisation fœtale où la rétine neurosensorielle, plissée, avec un aspect histologique en pseudo-rosettes multicellulaires (par opposition aux rosettes de Flexner-Wintersteiner), est séparée de l'épithélium pigmentaire et collée au vitré primitif en arrière du cristallin, réalisant un tableau de « pseudo-gliome » [1]. Devant ce tableau, les diagnostics différentiels de la trisomie 13 sont la maladie de Norrie, le syndrome de Walker-Warburg et le syndrome de Meckel. Il existe généralement une microphtalmie variable (de minime à extrême). Les autres atteintes oculaires possibles sont : opacités cornéennes, colobomes, cataractes, dysplasies des nerfs optiques.
La trisomie 18, encore appelée syndrome d'Edwards, est souvent incompatible avec une survie prolongée du fait des malformations cardiaques associées. La plupart des décès ont lieu avant l'âge de 1 an. Les enfants présentent une dysmorphie faciale avec micrognathie, implantation anormale des sourcils et parfois microcéphalie.
Les mosaïques représentent 5 % des observations de trisomie 18. Le tableau clinique est variable. Certains enfants ont un phénotype proche de la normale, sans dysmorphie ni retard mental et compatible avec une survie longue. D'autres ont un phénotype peu différent du syndrome d'Edwards complet.
Les malformations oculaires sont rares dans la trisomie 18. Elles consistent le plus souvent en des plis rétiniens dans le cadre d'une dysplasie rétinienne. Plus rarement, des colobomes choriorétiniens ou du nerf optique ont été rapportés.
La trisomie 21 est caractérisée d'un point de vue génétique par l'existence d'un chromosome 21 surnuméraire. L'ensemble des mécanismes physiopathogéniques permettant de comprendre cette affection reste encore mal compris.
Une partie du phénotype de ces patients est due à la surexpression de certains gènes localisés sur l'ensemble de ce chromosome 21. En effet, certains gènes ont une expression qui représente 1,5 fois la normale, traduisant leur activation sur le chromosome surnuméraire. Il est probable que ces gènes participent au phénotype des patients trisomiques. D'autres ont une expression discrètement augmentée, alors qu'un troisième groupe de gènes n'est pas surexprimé. Ces derniers ne participent probablement pas au phénotype de ces patients.
Il existe des régions critiques, telle la région 21q22.12/21q22.13, au sein desquelles sont localisés des gènes impliqués dans certains traits phénotypiques de ces patients. La surexpression du gène DYRK1A (qui code pour l’enzyme Dual specificity tyrosine-phosphorylation-regulated kinase 1A), localisé dans cette région d'intérêt, associée à celle du gène DSCR1 (down region critical region), également localisé dans cette région du chromosome 21, aboutit à une dérégulation de différentes voies de signalisation du fait de l'inhibition ou de l'altération de la réponse de différents facteurs de transcription, tels que NFATc (nuclear factor of activated T-cells), impliqué dans le développement des vertébrés dans leur ensemble, ou NRSF/REST (neuron-restrictive silencer factor/RE1-silencing transcription factor). La dérégulation de ce dernier entraîne des conséquences pathologiques précoces et sévères, perturbant le développement des différentes lignées tissulaires de l'embryon.
D'autres mécanismes de dérégulation de l'expression des gènes sont également retrouvés. Il est suspecté que la surexpression d'un ou plusieurs gènes du chromosome 21 entraîne des modifications de la chromatine nucléaire, aboutissant à des perturbations du transcriptome au sein des cellules de ces patients. Enfin, l'expression du facteur de transcription AIRE (auto-immune regulator), localisé sur le chromosome 21, est réduite dans le thymus de ces patients, malgré la présence d'un chromosome surnuméraire. Cette altération permet d'expliquer la prédisposition aux pathologies auto-immunes observées.
Le phénotype des patients porteurs d'une trisomie 21 relève donc d'un ensemble de dérégulations de facteurs de transcription et de leurs voies de contrôle, perturbant le développement des différents tissus tout au long de l'embryogenèse.
Le tableau clinique associe variablement : dysmorphie (fentes palpébrales dites mongoloïdes, épicanthus, nuque plate, visage arrondi, petit nez, pli palmaire unique bilatéral, petite taille); malformations (cardiaque : canal atrioventriculaire, digestive : atrésie duodénale); déficit intellectuel variable; épilepsie; maladie de Hirschsprung; fréquence accrue des leucémies et des cancers solides, des affections auto-immunes et endocrines; vieillissement précoce.
Une meibomite chronique est présente chez la moitié des patients. Elle entraîne une sécheresse oculaire, des chalazions récidivants, et favorise les frottements oculaires, eux-mêmes impliqués dans la physiopathologie du kératocône. Elle doit donc être systématiquement recherchée et traitée.
L'obstruction lacrymonasale est très fréquente et entraîne souvent un épiphora. Ses causes sont multiples : imperforation de la valvule de Hasner, agénésie des points lacrymaux, stricturotomie, atrésie du canalicule, mais surtout étroitesse du nez avec œdème de la muqueuse nasale à l'occasion des fréquents épisodes de rhinite, avec larmoiement et stase lacrymale très fréquents dans la petite enfance. La prise en charge est donc adaptée à chacune de ces situations en fonction du mécanisme suspecté.
Les syndromes du strabisme précoce sont rares; les ésotropies accommodatives sont en revanche très fréquentes si elles ne sont pas prévenues par le port de la correction optique totale dans l'enfance [2].
Les nystagmus sont présents chez environ un quart des enfants; on rencontre les trois principales variétés de nystagmus : syndrome du nystagmus précoce; nystagmus de type latent; nystagmus de type spasmus nutans [3].
Les troubles de la réfraction sont la règle dans la trisomie 21 : hypermétropie (36 % ), myopie (64 % ), astigmatisme (64 % ) [4]. L'accommodation des enfants avec trisomie 21 est retrouvée dans environ 75 % des cas. Cette parésie voire paralysie de l'accommodation doit donc être systématiquement recherchée et prise en charge par le port de verres à double foyer puis progressifs, qui permettront aux enfants de voir net de près et de ne pas aggraver ainsi leurs difficultés scolaires [5]. La skiascopie dynamique était une technique utilisée au xx siècle permettant d'évaluer cliniquement l'accommodation et ce même chez les tout petits, mais elle n'est plus d'actualité dans la pratique courante. Ce trouble de l'accommodation explique aussi pourquoi le « seuil de correction » de l'hypermétropie (degré d'hypermétropie justifiant d'une correction optique) doit être très bas chez ces enfants. Cette parésie de l'accommodation, constitutive de la trisomie 21, a été historiquement attribuée à l'atropine qui avait été instillée pour réaliser des cycloplégies. C'est pourquoi l'atropine a été longtemps réputée – à tort – contre-indiquée dans la trisomie 21. L'usage d'une cycloplégie est au contraire essentiel, car la parésie de l'accommodation est inconstante et souvent partielle, tandis que le port de la correction optique totale – ou même chez le tout petit « surtotale » – est indiqué [6]. Le choix du collyre cycloplégiant est en revanche guidé par l'âge de l'enfant et tient compte des affections possiblement associées, cérébrales ou cardiaques.
1. Le lecteur trouvera de plus amples informations sur la skiascopie en consultant les liens Internet suivants : https://www.larefraction.net/Telechargement/TelechTexte/files/Skiascopie.pdf, http://www.larefraction.net/Documents/Ref-Enfant/RefEnft_Skiascopie/RefEnft_Skiascopie.html
Le kératocône est rare chez l'enfant; il apparaît généralement à l'adolescence ou chez l'adulte, où sa prévalence est comprise entre 3 et 20 % . Il est très probablement favorisé par les frottements itératifs des paupières, qui sont prévenus par le traitement précoce des blépharites chroniques et le port de la correction optique. Sa prise en charge est d'autant plus difficile que la déficience intellectuelle est importante.
Les taches de Brushfield sont présentes chez environ trois quarts des patients avec trisomie 21 (contre un quart des cas dans la population générale). Elles résultent en fait d'une hypoplasie du stroma irien situé autour de la tache, elle-même constituée de tissu irien normal ou hypercellulaire.
La fréquence des cataractes congénitales ou précoces (environ 5 % ) justifie un examen ophtalmologique systématique des nourrissons avec un diagnostic de trisomie 21 dans les premiers mois de vie. Les cataractes blanches obturantes sont chirurgicales, contrairement aux cataractes céruléennes. Un lenticône ou un lentiglobe bilatéral peuvent parfois être observés dans la trisomie 21 (il s'agit d'une déformation du cristallin respectivement conique ou hémisphérique).
Il existe des hyperplasies de l'épithélium pigmentaire de la rétine chez environ un tiers des enfants avec trisomie 21 [7].
La morphologie de la papille optique dans la trisomie 21 est particulière dans environ 70 % des cas : la division des vaisseaux se fait très précocement, de sorte que le nombre de vaisseaux émergeant des limites du disque est excessif [8]. Dans certains cas, cette dysgénésie vasculaire s'accompagne d'un pseudo-œdème papillaire caractéristique, qui ne doit pas être confondu avec un œdème de stase vrai.
L'épaisseur fovéale est plus importante chez les enfants avec trisomie 21 que dans la population générale, ce qui suggère un développement fovéal anormal dans cette pathologie [9].
Le syndrome de Turner est dÛ à l'absence du second chromosome sexuel. Il s'agit d'une monosomie X avec un caryotype 45,X (dans les formes en mosaïque, le caryotype est 45,X/46,XX ou 45,X/46,XY). Le phénotype est féminin; les patientes atteintes n'ont qu'un chromosome X. Elles ont néanmoins une carence en œstrogènes et sont stériles. Il est constamment retrouvé une dysmorphie avec une micrognathie, une petite taille, un cou palmé, une implantation anormale des cheveux, des oreilles mal ourlées, un palais étroit, une poitrine large et un cubitus valgus. Mais d'autres anomalies congénitales du visage et des oreilles ont été rapportées. L'âge osseux est retardé avec une diminution de la minéralisation osseuse. Il existe un risque accru de malformations cardiaques et urinaires. Enfin, ces patientes développent volontiers des infections de l'oreille moyenne, des pathologies cardiovasculaires (notamment une hypertension artérielle), hépatiques (élévation des enzymes hépatiques ou cirrhose du foie) ou endocriniennes (hypothyroïdie, diabète, etc.).
Les patientes ont des capacités intellectuelles normales et une aptitude verbale préservée. En revanche, elles peuvent présenter des déficits dans certaines tâches dites « visuo-spatiales » et dans les interactions sociales.
Dans les formes en mosaïque, il existe souvent des dysgénésies du segment antérieur avec un risque de glaucome [10].
[1] Reese AB, Blodi FC Retinal dysplasia Am J Ophthalmol ( 1950 ) : 33: 23-32 illust
[2] Haugen OH, Hovding G Strabismus and binocular function in children with Down syndrome. A population-based, longitudinal study Acta ophthalmologica Scandinavica ( 2001 ) : 79: 133-139
[3] Wagner RS, Caputo AR, Reynolds RD Nystagmus in Down's syndrome Ophthalmology ( 1990 ) : 97: 1439-1444
[4] Cregg M, Woodhouse JM, Pakeman VH Accommodation and refractive error in children with Down syndrome : cross-sectional and longitudinal studies Invest Ophthalmol Vis Sci ( 2001 ) : 42: 55-63
[5] Stewart RE, Margaret Woodhouse J, Trojanowska LD In focus : the use of bifocal spectacles with children with Down's syndrome Ophthalmic Physiol Opt ( 2005 ) : 25: 514-522
[6] Parsa CF, Adyanthaya R Why atropine drops should be used in Down syndrome Br J Ophthalmol ( 2008 ) : 92: 295-296
[7] Stirn Kranjc B Ocular abnormalities and systemic disease in Down syndrome Strabismus ( 2012 ) : 20: 74-77
[8] Sherk MC, Williams TD Disc vascularity in Down's syndrome Am J Optom Physiol Opt ( 1979 ) : 56: 509-511
[9] O'Brien S, Wang J, Smith HA Macular structural characteristics in children with Down syndrome Graefes Arch Clin Exp Ophthalmol ( 2015 ) : 253: 2317-2323
[10] Lloyd IC, Haigh PM, Clayton-Smith J Anterior segment dysgenesis in mosaic Turner syndrome Br J Ophthalmol ( 1997 ) : 81: 639-643
G. Le Meur
Des pathologies de l'appareil rénal et urogénital peuvent avoir un lien avec l'ophtalmologie en associant, aux atteintes rénales ou des voies urinaires, une atteinte des divers tissus oculaires. Nous décrirons ici certains syndromes qui présentent notamment une atteinte cornéenne, cristallinienne ou rétinienne. Pour certaines pathologies, le lecteur pourra se référer au chapitre 26.7 sur les maladies métaboliques.
La cystinose est une maladie héréditaire, à transmission autosomique récessive (gène CTNS en position 17p13.2), qui est liée à un défaut de transport de la cystine hors des lysosomes. Ceci entraîne une accumulation lysosomale de cet acide aminé dans différents organes. La cystine accumulée induit la formation de cristaux caractéristiques au sein de nombreux tissus : les reins, les yeux, la moelle osseuse, le foie, la rate, le pancréas, la thyroïde, les muscles et le cerveau [1]. La prévalence est estimée à 1 sur 26 000 naissances en France [1]. Il existe trois formes cliniques : la cystinose néphropathique infantile (la plus fréquente), la cystinose juvénile et la cystinose bénigne oculaire [1]. L'atteinte oculaire, qui commence dès l'enfance sauf pour la cystinose bénigne oculaire où elle démarre plus tard, entraîne, dans la première décennie, un larmoiement et une photophobie en raison de l'accumulation de cristaux dans la conjonctive et la cornée. Ces dépôts apparaissent lors de l'examen à la lampe à fente comme une myriade de petits dépôts blancs brillants, réfringents (fig. 26-1) [2]. Le dépôt des cristaux commence dans la cornée périphérique, progressant de manière centripète avec l'âge [3]. Il a également été décrit une atteinte rétinienne avec l'apparition de zones de dépigmentation et des dépôts pigmentés rétiniens à plus long terme [4]. La formulation orale de cystéamine ne semble pas avoir un effet sur les cristaux de cystine cornéens [2]. En France, le Cystadrops 0,55 (Orphan Europe, Paris, France) peut être utilisé avec 4 instillations par jour, chez les enfants âgés de plus de 2 ans, mais uniquement par prescription hospitalière dans le cadre d'une autorisation temporaire d'utilisation (ATU) [5].
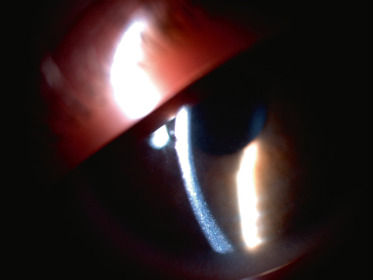
Fig. 26-1 Cystinose cornéenne.
La maladie de Fabry est une maladie héréditaire de surcharge lysosomale liée à la déficience de l'activité de l'alpha-galactosidase A lysosomale secondaire à des mutations dans le gène GLA(Xq21.3-q22) qui code pour l'enzyme alpha-galactosidase A. Les hommes et les femmes peuvent être atteints. Sa fréquence est estimée à 1 sur 40 000 à 117 000 naissances [6]. C'est une maladie progressive, multisystémique, d'expression variable, qui touche le cerveau, le cœur, les vaisseaux, les reins, les poumons, la peau et le tube digestif [6]. Trois signes oculaires sont associés : la cornea verticillata (fig. 26-2), la présence de vaisseaux conjonctivaux et/ou rétiniens tortueux et la cataracte sous-capsulaire [7]. Les signes ophtalmologiques ne sont retrouvés que dans environ 55 % des cas avec de manière plus fréquente l'atteinte cornéenne [7]. Ces dépôts verticillés sont présents dans la partie inférieure de la cornée au niveau de l'épithélium et du stroma antérieur. Une enzymothérapie substitutive utilisant une alpha-galactosidase A recombinante est actuellement utilisée.
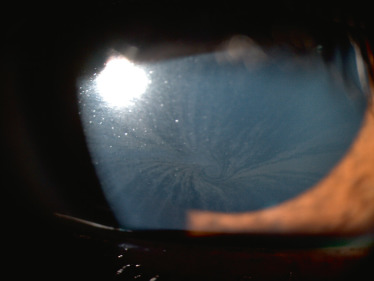
Fig. 26-2 Cornée verticillée.
Le syndrome d'Alport est une maladie génétique marquée par la survenue d'une néphropathie glomérulaire avec hématurie, puis d'une insuffisance rénale progressive associée à une surdité de perception ainsi qu'à des anomalies ophtalmologiques [8]. Sa fréquence est de 1 sur 10 000. Son mode de transmission est dans 85 % des cas lié à l'X avec une mutation du gène COL4A5 et dans 15 % des cas autosomique récessif avec une mutation des gènes COL4A3 ou COL4A4 [8]. De rares cas de transmission autosomique dominante ont été décrits [8]. Les anomalies ophtalmologiques surviennent par absence du collagène IV au niveau des membranes basales de la cornée, de la capsule cristallinienne ou de la rétine. Les manifestations ophtalmologiques sont des opacités cornéennes liées à des érosions récidivantes, un lenticône antérieur ou postérieur, une rétinopathie maculaire avec mouchetures ou flecks, un amincissement de la rétine temporale ou la survenue d'un trou maculaire [9]. Le risque de manifestations ophtalmologiques est estimé de 30 à 50 % pour la rétinopathie et de 15 à 30 % pour le lenticône, avec une fréquence plus importante chez les hommes [8]. Le diagnostic est important car la mise en place d'un traitement par inhibiteurs de l'enzyme de conversion (IEC) et antagonistes des récepteurs de l'angiotensine II permet de retarder l'apparition de l'insuffisance rénale [10].
Le syndrome de Lowe ou syndrome oculo-cérébro-rénal est un syndrome multiviscéral rare, où est présente la triade clinique : cataracte bilatérale, déficience intellectuelle et dysfonction tubulaire rénale proximale [11]. La maladie, de transmission récessive liée au chromosome X, est due à une mutation du gène OCRL1, situé en Xq26.1 qui code pour une phosphatidylinositol 4,5-bisphosphate-5-phosphatase [12]. Sa prévalence est estimée à 1 sur 50 0000 dans la population générale [11]. Dès la naissance, une hypotonie sévère ainsi qu'une cataracte bilatérale dense sont retrouvées puis, dans les mois qui suivent, une tubulopathie rénale proximale apparaît. Un peu plus tard dans l'enfance sont notés un retard psychomoteur et un retard de croissance. Des troubles du comportement ainsi qu'une insuffisance rénale marquent l'adolescence [13]. De nombreux patients développent, en plus, une arthropathie invalidante, des nodules sous-cutanés ou une aréflexie. Au niveau ophtalmologique, un glaucome sévère peut survenir la première année voire dans les deux premières décennies. La cataracte est liée à une dégénérescence des fibres primitives postérieures du cristallin embryonnaires. Une atteinte cornéenne avec des cicatrices chéloïdes sans notion de traumatisme peut survenir chez 25 % des enfants avant l'âge de 5 ans. L'acuité visuelle est rarement supérieure à 20/100 notamment à cause d'une atteinte rétinienne primitive [11]. La prise en charge comprend une chirurgie précoce de la cataracte avec une correction de l'aphaquie par lunettes s'il n'y a pas eu mise en place d'un implant intra-oculaire; l'implantation est classiquement discutée chez ces patients fragiles (même si désormais la chirurgie de la cataracte est moins pourvoyeuse d'inflammation et de cataractes secondaires qui requièrent des anesthésies itératives chez le petit enfant); l'adaptation en lentilles de contact est difficile à cause de la fragilité cornéenne [11]. Le traitement est symptomatique et l'espérance de vie dépasse rarement la quarantaine.
Le syndrome WAGR (Wilms tumor, aniridia, genital anomalies, mental retardation) associe tumeur de Wilms, aniridie, anomalies génito-urinaires et retard mental [14]. Le terme aniridie (voir chapitre 11) désigne en fait un ensemble de malformations oculaires complexes impliquant notamment le limbe, l'iris et la fovéa, La prévalence du syndrome WAGR est inférieure à 1 pour 100 000 personnes. Le syndrome est la conséquence d'une microdélétion de la région 11p13 du chromosome 11 englobant les gènes WT1 et PAX6 [15, 16]. La microdélétion survient de novo dans la plupart des cas. La tumeur de Wilms, ou néphroblastome, est la tumeur rénale de l'enfant la plus fréquente et peut survenir à tout âge [17]. À celle-ci, qui se développe entre 42,5 et 77 % des cas en fonction de la grandeur de la délétion [18], peuvent être associés une aniridie totale ou partielle avec éventuellement une cataracte ou un glaucome, des anomalies génito-urinaires allant de l'ambiguïté sexuelle à l'ectopie testiculaire, ainsi qu'un déficit intellectuel de degré variable. Le risque qu'une aniridie sporadique entre dans le cadre d'un syndrome WAGR est de 30 % ; il est donc essentiel de rechercher ce syndrome devant tout nouveau cas sporadique d'aniridie [19]. Les patients porteurs d'un syndrome WAGR doivent bénéficier d'une surveillance trimestrielle par une échographie rénale jusqu'à leurs 6 ans au moins [18]. Outre le dépistage du néphroblastome par ces échographies régulières, la surveillance de la fonction rénale et la recherche d'une hypertension artérielle doivent être débutées dès l'enfance et se poursuivre tout au long de la vie, car le risque d'insuffisance rénale à 20 ans est estimé à 38 % [20].
Le syndrome rein-colobome (SRC) également appelé syndrome papillo-rénal est une anomalie génétique, de transmission autosomique dominante liée au gène PAX2, qui induit une dysplasie du nerf optique et une hypodysplasie rénale [21]. Des signes oculaires sont présents dans 77 % des cas, des signes rénaux dans 92 % des cas et des signes auditifs dans 7 % des cas avec diminution de l'acuité auditive [22]. Les anomalies oculaires se traduisent, majoritairement, par une atteinte du nerf optique : nerf optique dysplasique, colobome papillaire ou morning glory (fig. 26-3) [21]. D'autres anomalies oculaires, comme un colobome rétinien, une microcornée, un staphylome scléral, un kyste du nerf optique, une microphtalmie, une hypoplasie fovéale ou une anomalie maculaire de pigmentation, ont été décrites [21]. Les malformations rénales (hypoplasie rénale, hypodysplasie rénale ou reins multikystiques) sont retrouvées, fréquemment avant la découverte des anomalies ophtalmologiques, en se manifestant par une hypertension artérielle, une protéinurie ou un reflux vésico-urétéral, voire une insuffisance rénale [21], Le lecteur peut se référer sur le sujet également au chapitre 21.
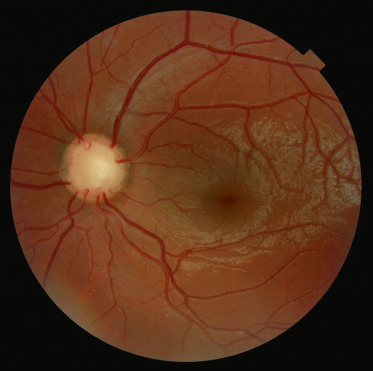
Fig. 26-3 Anomalie du nerf optique dans le cadre d’un syndrome oculorénal.
Les ciliopathies représentent un nouveau groupe de maladies génétiques causées par un dysfonctionnement de deux organites fonctionnellement et physiquement associés, le centrosome et le cil [23], Elles comprennent notamment le syndrome de Bardet-Biedl et les néphronophtises, dont le syndrome de Senior-Loken, La néphronophtise est une néphropathie tubulo-interstitielle chronique qui évolue vers l'insuffisance rénale terminale, Il y a trois formes de néphronophtises suivant l'âge de survenue de l'atteinte rénale : infantile, juvénile ou adolescente, C'est dans la forme juvénile que l'atteinte rétinienne est la plus fréquente, Une dystrophie rétinienne de type périphérique (rod-cone) est retrouvée lors d'une mutation des gènes NPHP3, NPHP4, NPHP5 et NPHP6 [24]. La description initiale du syndrome de Senior-Loken est l'association d'une atteinte dégénérative rétinienne sévère et précoce (à type d'amaurose congénitale de Leber) à une néphronophtise juvénile, Actuellement, le spectre d'atteinte rétinienne qui rentre dans ce syndrome va de l'atteinte sévère rétinienne à une atteinte modérée et plus tardive (fig. 26-4).
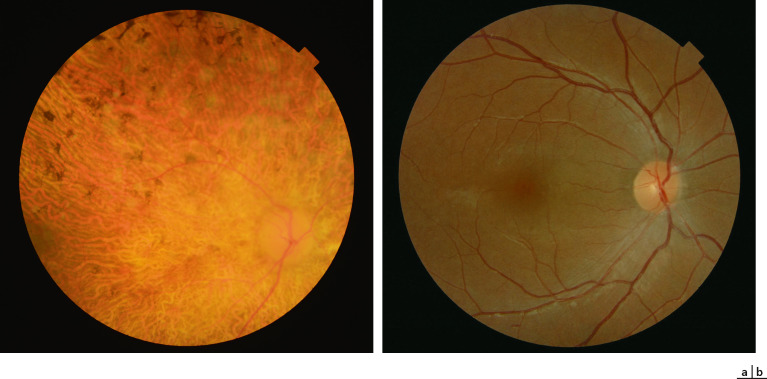
Fig. 26-4 Atteinte rétinienne associée à une néphronopthise.
a. Atteinte sévère avec ostéoblastes périphériques. b. Atteinte modérée avec maculopathie modérée.
[1] Elmonem MA, Veys KR, Soliman NA Cystinosis : a review Orphanet J Rare Dis ( 2016 ) : 11: 47 p
[2] Shams F, Livingstone I, Oladiwura D, Ramaesh K Treatment of corneal cystine crystal accumulation in patients with cystinosis Clin Ophthalmol Auckl NZ ( 2014 ) : 8: 2077-2084
[3] Tsilou E, Zhou M, Gahl W Ophthalmic manifestations and histopathology of infantile nephropathic cystinosis : report of a case and review of the literature Surv Ophthalmol ( 2007 ) : 52: 97-105
[4] Tsilou ET, Rubin BI, Reed G Nephropathic cystinosis: posterior segment manifestations and effects of cysteamine therapy Ophthalmology ( 2006 ) : 113: 1002-1009
[5] Labbé A, Baudouin C, Deschênes G A new gel formulation of topical cysteamine for the treatment of corneal cystine crystals in cystinosis : the Cystadrops OCT-1 study Mol Genet Metab ( 2014 ) : 111: 314-320
[6] Meikle PJ, Hopwood JJ, Clague AE, Carey WF Prevalence of lysosomal storage disorders JAMA ( 1999 ) : 281: 249-254
[7] Pitz S, Kalkum G, Arash L Ocular signs correlate well with disease severity and genotype in Fabry disease PloS One ( 2015 ) : 10: e0120814 p
[8] Hertz JM, Thomassen M, Storey H, Flinter F Clinical utility gene card for : Alport syndrome – update 2014 Eur J Hum Genet ( 2015 ) : 23:
[9] Savige J, Sheth S, Leys A Ocular features in Alport syndrome : pathogenesis and clinical significance Clin J Am Soc Nephrol ( 2015 ) : 10: 703-709
[10] Kashtan CE, Ding J, Gregory M Clinical practice recommendations for the treatment of Alport syndrome : a statement of the Alport Syndrome Research Collaborative Pediatr Nephrol Berl Ger ( 2013 ) : 28: 5-11
[11] Bokenkamp A, Ludwig M The oculocerebrorenal syndrome of Lowe : an update Pediatr Nephrol Berl Ger ( 2016 ) : 31: 2201-2212
[12] Attree O, Olivos IM, Okabe I The Lowe's oculocerebrorenal syndrome gene encodes a protein highly homologous to inositol polyphosphate-5-phosphatase Nature ( 1992 ) : 358: 239-242
[13] Loi M Lowe syndrome Orphanet J Rare Dis ( 2006 ) : 1: 16 p
[14] Miller RW, Fraumeni JF, Manning MD Association of Wilms's tumor with aniridia, hemihypertrophy and other congenital malformations N Engl J Med ( 1964 ) : 270: 922-927
[15] Muto R, Yamamori S, Ohashi H, Osawa M Prediction by FISH analysis of the occurrence of Wilms tumor in aniridia patients Am J Med Genet ( 2002 ) : 108: 285-289
[16] Xu S, Han JC, Morales A Characterization of 11p14-p12 deletion in WAGR syndrome by array CGH for identifying genes contributing to mental retardation and autism Cytogenet Genome Res ( 2008 ) : 122: 181-187
[17] Dumoucel S, Gauthier-Villars M, Stoppa-Lyonnet D Malformations, genetic abnormalities, and Wilms tumor Pediatr Blood Cancer ( 2014 ) : 61: 140-144
[18] Clericuzio C, Hingorani M, Crolla JA Clinical utility gene card for : WAGR syndrome Eur J Hum Genet ( 2011 ) : 19:
[19] Gronskov K, Olsen JH, Sand A Population-based risk estimates of Wilms tumor in sporadic aniridia. A comprehensive mutation screening procedure of PAX6 identifies 80 % of mutations in aniridia Hum Genet ( 2001 ) : 109: 11-18
[20] Breslow NE, Norris R, Norkool PA Characteristics and outcomes of children with the Wilms tumor-Aniridia syndrome : a report from the National Wilms Tumor Study Group J Clin Oncol ( 2003 ) : 21: 4579-4585
[21] Schimmenti LA Renal coloboma syndrome Eur J Hum Genet ( 2011 ) : 19: 1207-1212
[22] Bower M, Salomon R, Allanson J Update of PAX2 mutations in renal coloboma syndrome and establishment of a locus-specific database Hum Mutat ( 2012 ) : 33: 457-466
[23] Hurd TW, Hildebrandt F Mechanisms of nephronophthisis and related ciliopathies Nephron Exp Nephrol ( 2010 ) : 118: e9-14
[24] Ronquillo CC, Bernstein PS, Baehr W Senior-Løken Syndrome : A syndromic form of retinal dystrophy associated with nephronophthisis Vision Res ( 2012 ) : 75: 88-97
C. Orssaud
L'œil et l'oreille sont parfois, du fait de certaines similitudes embryologiques ou histologiques, affectés par un même processus, qu'il soit polymalformatif (dysgénésique, comme le syndrome CHARGE, ou disruptif, comme la rubéole congénitale) ou dégénératif (comme dans les syndromes d'Usher, de Wolfram, Kearns-Sayre ou d'Alport). Il est absolument fondamental pour l'ophtalmologiste, devant certains tableaux plus ou moins spécifiques, de s'assurer de la normalité de la fonction auditive, afin que soient alors pris en charge au plus vite et de façon concertée les deux aspects de ce double handicap, que tout retard au diagnostic est susceptible d'aggraver. Les différentes caractéristiques des principaux syndromes oculo-auditifs sont récapitulées dans le tableau 26-1.
Tableau 26-1 – Les différentes caractéristiques des principaux syndromes oculo-auditifs.
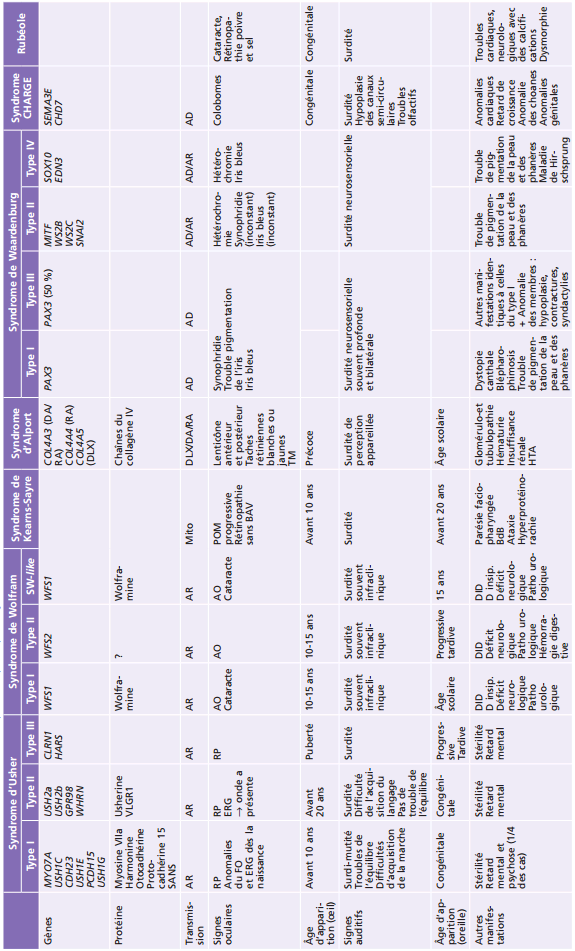
AD : autosomique dominant ; AO : atrophie optique ; AR : autosomique récessif ; BAV : baisse d’acuité visuelle ; BdB : bloc de branche ; D insip. : diabète insipide ; DID : diabète insulino-dépendant ; DLX : dominant liée au chromosome X ; CHARGE : Coloboma, Heart defect, Atresia choanae, Retarded growth, Genital anomalies, Ear anomalies ; ERG : électrorétinogramme ; FO : fond d’oeil ; HTA : hypertension artérielle ; Mito : mitochondrial ; POM : paralysie oculomotrice ; RP : rétinopathie pigmentaire ; SW : syndrome de Wolfram ; TM : trou maculaire.
G. Le Meur, C. Orssaud, M. Robert
L'appellation « syndromes cutanés » désigne parfois un groupe de syndromes, d'associations et de maladies variées, aux limites floues, dont l'œil constitue toujours évidemment, du fait de ses origines embryologiques, un élément central.
Les phacomatoses sont des affections génétiques neurocutanées, parfois aussi appelées dysplasies neuro-ectodermiques ou neurocristopathies. Elles sont classiquement divisées en trois grands sous-groupes : les phacomatoses classiques (neurofibromatoses de type 1 et 2; sclérose tubéreuse de Bourneville; syndromes de von Hippel-Lindau, de Protée, de Cowden et du nævus épidermique), les phacomatoses vasculaires (syndromes de Sturge-Weber-Krabbe, de Fabry, de Rendu-Osler et ataxie-télangiectasie) et les phacomatoses pigmentaires (syndromes de Waardenburg, incontinentia pigmenti et hypomélanose d'Ito). La notion même de phacomatose, qui résulte de considérations historiques, est actuellement débattue et peu à peu abandonnée au profit de celle d'hamartomatose, plus restrictive. Le plan adopté ici privilégie volontairement les organes affectés au détriment de classifications anciennes fondées sur des hypothèses physiopathologiques non avérées.
La neurofibromatose de type 1 (NF1), anciennement appelée maladie de Recklinghausen, est une maladie génétique neurocutanée, cliniquement hétérogène, qui associe la présence de neurofibromes bénins et de tumeurs malignes du système nerveux central et périphérique. Sa fréquence est de 1/3000 à 1/4000 dans le monde. Elle est marquée par une prédisposition à l'apparition de tumeurs. Les manifestations cliniques sont extrêmement variables avec une grande hétérogénéité allant d'une atteinte de forme minime, peu visible, à des formes graves qui peuvent mettre en jeu le pronostic vital. Dans la moitié des cas de NF1, les signes cliniques apparaissent dès la première année et dans 97 % avant l'âge de 8 ans [1]. Les critères de diagnostic sont présentés dans l’encadré 26-1. Les formes graves ne représentent qu'environ 15 % des cas.
2. Sous-partie rédigée par G. Le Meur.
Critères de diagnostic de neurofibromatose de type 1
Le diagnostic est porté si deux des sept critè res suivants sont présents (é tablis lors de la confé rence de consensus de Bethesda aux États-Unis en 1988) :
un apparenté de premier degré atteint (parent, frè re, soeur ou enfant) ;
au moins 6 taches café au lait de diamè tre supé rieur à 1,5 cm aprè s la puberté ou supé rieur à 0,5 cm avant la puberté ;
la pré sence de lentigines axillaires ou inguinales ;
au moins deux neurofibromes quel que soit leur type ou un neurofibrome plexiforme ;
un gliome des voies visuelles ;
au moins deux nodules de Lisch (ou hamartomes iriens) ;
une lé sion osseuse caracté ristique (pseudarthrose, dysplasie du sphé noï de, ou amincissement du cortex des os longs).
La neurofibromatose de type 1 est une maladie de transmission autosomique dominante à pénétrance complète. Le gène NF1, découvert en 1990, est un gène de grande taille (61 exons) localisé en 17q11.22 qui code pour une protéine cytoplasmique de 327 kDa appelée neurofibromine. Cette protéine est une protéine suppresseur de tumeur, qui joue un contrôle négatif pour le proto-oncogène RAS en diminuant les signaux mitogènes [2]. Environ la moitié des personnes atteintes de NF1 n'ont aucune histoire familiale de neurofibromatose de type 1, car ce gène est fréquemment atteint de mutation spontanée ou dite « de novo » . Un effet pléiotropique est également décrit pour cette pathologie : des patients avec la même mutation, au sein d'une même famille peuvent avoir des signes cliniques totalement différents suggérant un rôle de gènes modificateurs ou de phénomènes épigénétiques responsables de la variabilité de l'expression phénotypique de la NF1 [1]. Il ne semble pas y avoir de corrélation phénotype-génotype. Toutefois, les personnes atteintes de la mutation spécifique c.2970-2972 delAAT présentent des symptômes modérés de la pathologie [3], alors que les patients atteints d'une microdélétion 17q11.2 présentent des neurofibromes étendus avec dysmorphie faciale et des difficultés d'apprentissage [4, 5]. Actuellement, le diagnostic de neurofibromatose de type 1 est établi sur des critères cliniques, réservant le diagnostic génétique à des cas non typiques ou à des demandes de conseil génétique.
Au niveau cutané, certaines modifications, qui apparaissent précocement, caractérisent cette maladie : taches café au lait, lentigos ou éphelides sur les aisselles et sur la région inguinale, autres anomalies comme les neurofibromes qui apparaissent plus tardivement.
Les taches café au lait, qui sont des taches pigmentées de couleur marron clair sans relief, sont une manifestation précoce et fréquente de la NF1. La présence de plus de 6 taches café au lait de taille> 0,5 cm chez une personne en âge prépubère ou de taille> 1,5 cm chez un individu post-pubère font partie des critères diagnostiques et doivent être un signe d'alerte pour les pédiatres (fig. 26-5a). Ces modifications pigmentaires cutanées n'ont pas de pouvoir dégénératif.
La présence d'éphélides ou lentigos (lentigines) axillaires ou inguinales apparaît également entre 5 et 8 ans. Ces lésions peuvent être présentes également au niveau d'autres plis cutanés du corps. Les éphélides ( « taches de rousseur » sont en théorie des taches plus planes que les lentigos qui sont des macules plus surélevées).
Trois types de neurofibromes peuvent être décrits. Les neurofibromes cutanés, indolores et de consistance molle, sont situés sur la peau et se développent après la puberté (fig. 26-5b). Les neurofibromes sous-cutanés sont situés sous la peau. Les neurofibromes plexiformes sont des neurofibromes mixtes (cutané et sous-cutané), de consistance molle avec des boules et des cordons fibreux durs, présents dès la naissance, souvent de manière unique soit sur le tronc, les membres ou la paupière, et pouvant dégénérer en tumeur maligne.
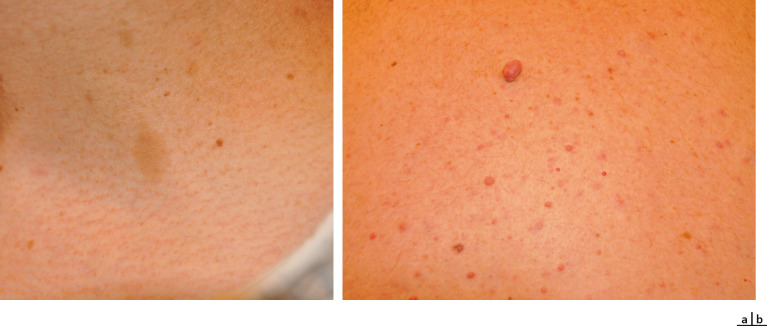
Fig. 26-5 Taches café au lait.
Les nodules de Lisch sont des hamartomes iriens pigmentés qui se rencontrent typiquement après l'âge de 6 ans (présents chez 15 à 20 % des enfants à l'âge de 6 ans et chez 95 % des adultes) (fig. 26-6) [6]. La présence de ces nodules est asymptomatique mais est un critère de diagnostic clinique et nécessite donc l'examen minutieux de l'iris en lampe à fente.
Le gliome des voies visuelles est une tumeur bénigne du nerf optique, du chiasma et/ou des bandelettes optiques, qui peut mettre un jeu le pronostic fonctionnel visuel (fig. 26-7) et constitue un autre critère de la maladie. Généralement, il s'agit d'un astrocytome pilocytique de bas grade. Dans le monde, sa fréquence est estimée entre 15 et 20 % des patients atteints d'une NF1 [2, 7]. Une étude récente, à propos d'une cohorte française, retrouve une fréquence de 14,7 % de gliomes des voies visuelles chez des patients ayant eu une imagerie par résonance magnétique (IRM) à titre systématique lors du diagnostic de NF1. De cette étude, nous savons que le gliome ne sera symptomatique que chez 20 % des patients porteurs d'un gliome au moment de la pose du diagnostic de NF1 et que sur l'ensemble des NF1, seuls 5 % des patients présenteront un gliome qui deviendra symptomatique [8]. L'âge de survenue du gliome est en moyenne de 4,2 ans mais la majorité des cas problématiques le sont avant l'âge de 6 ans. Les signes d'évolutivité sont la baisse d'acuité visuelle, les atteintes du champ visuel, les anomalies du réflexe pupillaire, l'œdème papillaire, l'atrophie optique, le strabisme, l'exophtalmie dans le cas d'une atteinte antérieure. Les trois signes les plus fréquents, par ordre de fréquence, sont la baisse de l'acuité visuelle, l'atteinte du nerf optique et l'exophtalmie. Dans cette étude, quand la première IRM réalisée chez les enfants lors du diagnostic était normale, cela est demeuré ainsi lors de l'examen 2 ans plus tard. L'atteinte tumorale peut survenir au niveau soit du nerf optique, soit du chiasma ou en rétrochiasmatique. Lorsque le gliome affecte le chiasma, il peut être associé à des troubles hormonaux, dont une puberté précoce. En cas d'indication à un traitement, le traitement recommandé, actuellement depuis 2004, est l'association carboplatine-vincristine en chimiothérapie de première intention. La radiothérapie, chez ces enfants qui ont une susceptibilité tumorale augmentée, n'est pas recommandée. Certaines publications montrent que les gliomes postérieurs ont une évolution plus péjorative que les gliomes antérieurs du nerf optique. Lorsque le gliome devient symptomatique, le pronostic fonctionnel reste alors réservé : lors du suivi des patients de la cohorte française sur plus de 7 ans, 30 % ont présenté une atteinte visuelle moyennement sévère à sévère et dans une étude multicentrique récente le devenir visuel des enfants à la fin des cycles de chimiothérapie pour gliome des voies visuelles montre 32 % d'amélioration, 40 % de stabilité et 32 % de dégradation [9, 10]. Le groupe d'experts français recommande une surveillance annuelle ophtalmologique chez tous les enfants ayant une NF1 avec un examen de l'acuité visuelle, une analyse du réflexe pupillaire, un champ visuel et un fond d'œil. L'indication de l'IRM systématique est réservée aux enfants dont le diagnostic de NF1 est fait avant l'âge de 6 ans, en raison de la difficulté d'obtenir un examen ophtalmologique complet avec champ visuel, ou aux enfants chez lesquels un trouble du comportement rend l'examen ophtalmologique aléatoire et difficile [9]. L'âge de fin de la surveillance n'a cessé d'augmenter au cours des dernières années : il était initialement de 6 ans, puis est passé à l'âge de 8 ans pour le National Institute of health (NIH) aux États-Unis, tandis que de nombreuses équipes européennes recommandent désormais un suivi semestriel jusqu'à 8 ans, puis un suivi annuel jusqu'à 18 ans en raison de l'existence de rares gliomes de début tardif.
Le neurofibrome plexiforme de la paupière (autre critère diagnostique) occupe généralement la paupière supérieure de manière unilatérale (fig. 26-8). C'est une tumeur qui provoque un ptosis avec une déformation en S de la paupière et une sensation de pelote de ficelle à la palpation. La fréquence du névrome plexiforme de la paupière supérieure varie de 1,5 à 17,6 % . Il est souvent associé à d'autres manifestations orbitofaciales dont les plus fréquentes sont l'hypertrophie de l'hémiface, le gigantisme orbitaire ou la dysplasie sphéno-orbitaire, affectant l'aile du sphénoïde (fig. 26-9), toutes étant des manifestations homolatérales. On parle de syndrome de François devant l'association de ces signes. Au niveau ophtalmologique, il peut être responsable d'amblyopie, de strabisme, d'anisométropie et de ptosis [11]. Il peut aussi s'associer, dans environ la moitié des cas chez l'enfant, à un glaucome congénital homolatéral. Plus rarement, il s'agit d'une myopie forte avec buphtalmie sans glaucome, le plus souvent dans le cadre d'un gigantisme orbitaire.
Les spécificités du glaucome de la NF1 sont traitées dans le chapitre 12. La physiopathologie fait probablement intervenir une endothélialisation progressive de l'angle iridocornéen, responsable à la fois du glaucome et de l'ectropion irien. Il est important de rechercher une NF1 devant un glaucome congénital unilatéral associé à un ectropion irien; il est aussi important de surveiller la biométrie, la pression intra-oculaire et la papille optique des petits enfants suivis pour NF1, particulièrement en cas d'ectropion irien ou de neurofibrome plexiforme de la paupière supérieure.
D'autres signes rétiniens, de description plus récente, sont d'une aide utile au diagnostic en raison de leur précocité d'apparition, mais ne constituent pas actuellement des critères de la maladie : il s'agit :
- –des taches hyperréflectives visibles sur les clichés infrarouges de la rétine, souvent réalisables dès l'âge de 2 à 3 ans (fig. 26-10), dont il a été montré sur une population mixte (adultes et enfants) qu'avec un cut-off à « 1,5 tache ou plus » , leur présence avait une sensibilité de 83 % et une spécificité de 96 % [12];
- –des anomalies vasculaires généralement uniques, isolées et unilatérales, d'une petite veinule rétinienne de deuxième ou troisième ordre, à type de tortuosité en tire-bouchon, présentes chez un tiers des patients (fig. 26-11) [13].
Les hamartomes astrocytaires et les ischémies rétiniennes sont très rares dans la NF1.
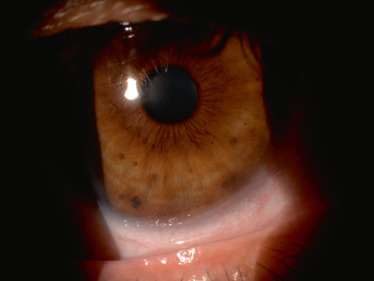
Fig. 26-6 Nodules de Lisch, apparaissant sous la forme de petites formations arrondies de couleur chamois et aux contours nets.
La lésion plane plus sombre aux contours spiculés, inférieure, est un banal nævus irien.
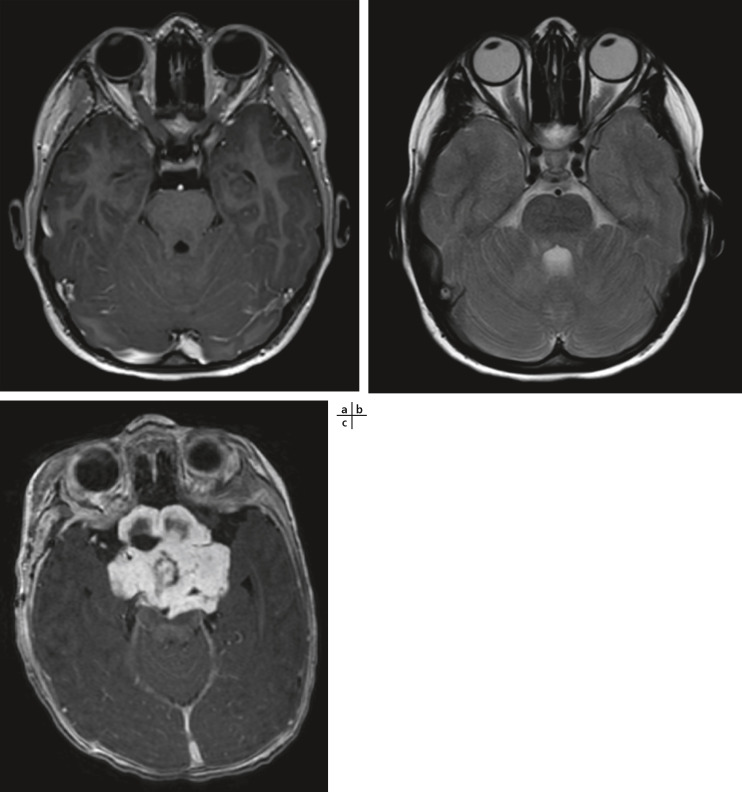
Fig. 26-7 Examen IRM de gliomes des voies visuelles.
La lésion plane plus sombre aux contours spiculés, inférieure, est un banal nævus irien.
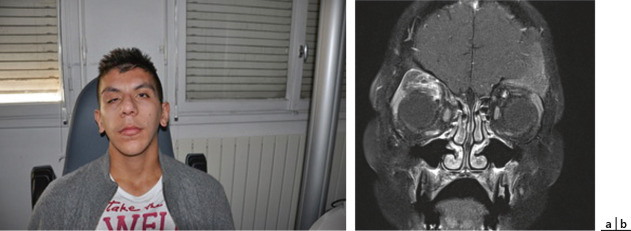
Fig. 26-8 Névrome plexiforme de la paupière supérieure droite.
a. Aspect clinique. b. Associé à une dysplasie de l’aile du sphénoïde homolatérale avec ectasie durale ; aspect en IRM : axial T1, axial T2 et coronal T1 SAT-FAT gadolinium.
(Fig. 26-8a : remerciements au Pr D. Denis.)
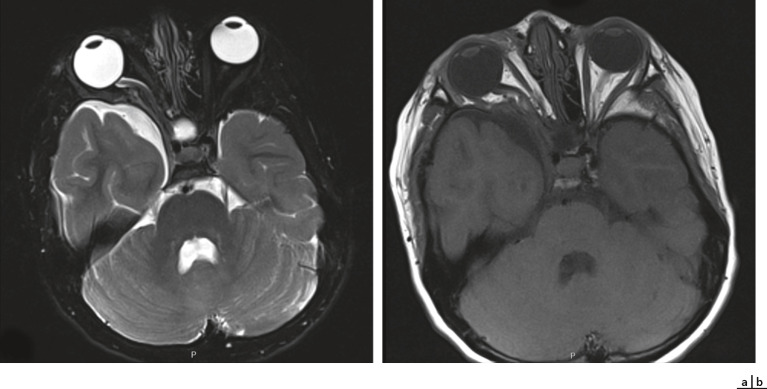
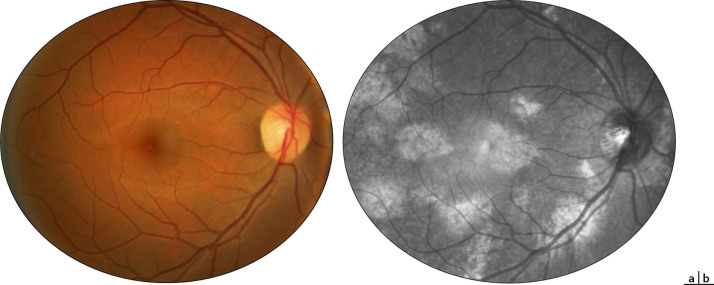
Fig. 26-10 Taches hyperréflectives de la NF1 invisibles ophtalmoscopiquement (a), mais visibles en imagerie en proche infrarouge (b).
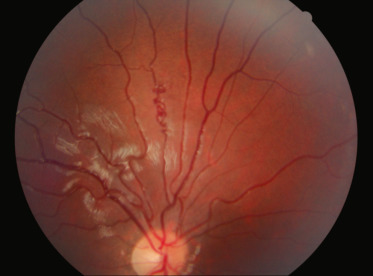
Fig. 26-11 Tortuosité en tire-bouchon d’une veine de deuxième ordre dans le cadre d’une NF1.
Les patients atteints de NF1 peuvent développer des anomalies squelettiques comme la scoliose, la dysplasie de l'aile du sphénoïde, la dysplasie frontale, la dysplasie congénitale du tibia, la pseudarthrose mais ils ont, aussi, une densité osseuse diminuée [14]. La présence d'ostéopathie n'est pas un facteur de risque pour la survenue de complications comme le gliome des voies visuelles, les neurofibromes plexiformes [15]. En revanche, le risque de fracture est cinq fois plus élevé chez les patients ayant une NF1 de plus de 40 ans et trois fois plus élevé dès l'âge de 16 ans [16]. La scoliose, qui est un des signes orthopédiques les plus fréquents, affecte 10 à 26 % des patients porteurs de NF1 et se manifeste vers l'âge de 10 ans [17]. Un examen annuel chez l'enfant et le jeune adolescent avec prise en charge orthopédique adaptée spécialisée doit être réalisé afin d'éviter les complications pulmonaires liées à la déformation thoracique [18].
La dysplasie des os longs est, quant à elle, diagnostiquée dès la première année de vie et affecte plus fréquemment le tibia dans le sens antérolatéral. Les autres atteintes osseuses peuvent être une croissance asymétrique des membres, la présence d'une pseudarthrose, habituellement du tibia [15]. La dysplasie unilatérale de l'aile du sphénoïde est fréquemment associée à un neurofibrome plexiforme ou à une ectasie durale respectivement dans 73,3 % et 80,0 % des cas [19]. Au niveau ophtalmologique, chez les patients qui présentent une dysplasie de l'aile du sphénoïde, il faut se méfier d'une amblyopie par anisométropie ou par neuropathie optique. Il est décrit une augmentation de 2 déviations standard (DS) du périmètre crânien chez les patients porteurs de NF1 [20]. Il est cependant probable que cette macrocéphalie résulte d'anomalies de développement des substances blanche et grise plus que de l'atteinte squelettique. Une macrocéphalie doit faire éliminer une hydrocéphalie, non exceptionnelle en cas de NF1 et secondaire à une compression par un gliome ou, plus souvent, à une sténose de l'aqueduc de Sylvius, cette sténose résultant d'une prolifération péri-aqueducale de cellules gliales sous-épendymaires.
L'atteinte neurologique est inconstante. Certains enfants atteints de NF1 présentent une déficience neuropsychologique spécifique impliquant notamment les habiletés motrices visuospatiales et fines, des troubles de la coordination motrice, un trouble des fonctions exécutives comme la mémoire de travail, la flexibilité cognitive et le contrôle inhibiteur [21]. Selon les études, environ 50 % des enfants atteints de NF1 ont des troubles d'apprentissage [22, 23] et environ 30 à 65 % répondent aux critères de diagnostic de trouble déficitaire de l'attention avec ou sans hyperactivité (TDAH) [24, 25]. Ces divers troubles peuvent entraîner des problèmes scolaires et des troubles des apprentissages chez plus de 40 % des enfants atteints de NF1 [21]. Les IRM cérébrales, chez environ 70 % des patients atteints de NF1, mettent en évidence des anomalies de signal de la substance blanche, appelées des objets brillants non identifiés (OBNI), situées en sous-cortical particulièrement dans les noyaux gris centraux, le thalamus, le cervelet et le tronc cérébral [26]. Les patients atteints de NF1 ont un risque accru de développer des tumeurs du système nerveux central et périphérique, notamment les neurofibromes plexiformes. Ces tumeurs bénignes des gaines des nerfs sont associées à : une morbidité et une mortalité augmentée; une diminution de la qualité de vie en raison de la défiguration; un risque de la compression des structures vitales; une douleur souvent chronique. La transformation maligne de ces lésions est parfois difficile à diagnostiquer : une modification de taille, un neurofibrome qui devient douloureux doivent faire craindre une transformation maligne.
Des neurofibromes profonds peuvent se développer dans l'espace médiastinal ou péritonéal induisant des signes cliniques spécifiques liés à la compression. Au niveau endocrinien, un phéochromocytome peut être observé. Les manifestations vasculaires sont surtout marquées par des rétrécissements artériels notamment de l'artère rénale ou des artères cérébrales comme les anomalies retrouvées dans la maladie de Moya Moya.
Actuellement, il n'y a pas de traitement qui ait d'autorisation de mise sur le marché (AMM) pour cette indication. Une prise en charge multidisciplinaire des complications de cette maladie est nécessaire avec des praticiens alertés aux divers problèmes rencontrés chez ces patients. Une prise en charge orthophonique et en psychomotricité ainsi que par un centre des troubles des acquisitions ou du langage peut être nécessaire quand des difficultés scolaires sont rencontrées. Un soutien psychologique peut s'avérer nécessaire. Dernièrement, le sirolimus, qui est un inhibiteur de mTOR (mammalian target of rapamycin), a été testé dans les neurofibromes avec une diminution des douleurs ressenties par les patients [27].
La neurofibromatose de type 2 (NF2) est une maladie autosomique dominante rare liée à une mutation du gène NF2 (22q12.2), qui code une protéine suppresseur de tumeur : la merline (ou schwannomine ou neurofibrimine 2). Sa fréquence est estimée à 1/60 000. La maladie est caractérisée par l'apparition progressive – le plus souvent à partir de 18 ans, rarement dès l'enfance – de schwannomes et de méningiomes. La seule présence de schwannomes vestibulaires bilatéraux, caractéristique, suffit à poser le diagnostic. Les patients atteints peuvent également développer des épendymomes ou rarement des astrocytomes. Il s'agit d'une maladie à critères; ceux-ci sont présentés dans l’encadré 26-2.
3. Sous-partie rédigée par G. Le Meur, M. Robert.
Critères de diagnostic de neurofibromatose de type 2
Diagnostic certain : schwannome bilatéral du VIII.
Diagnostic probable :
- antécédent familial de NF2 ;
- et schwannome vestibulaire unilatéral ou deux atteintes parmi les suivantes : méningiome, gliome, schwannome, cataracte sous-capsulaire postérieure, cataracte corticale juvénile.
En ophtalmologie, une cataracte sous-capsulaire est fréquemment retrouvée (60 à 80 % des cas) et est parfois le premier signe clinique avant l'apparition des schwannomes [28]. L'atteinte ophtalmologique la plus caractéristique est le méningiome des gaines du nerf optique, uni- ou bilatéral. Il doit être évoqué devant une atrophie optique d'importance variable, d'évolution très lente et insidieuse en sorte que la révélation de l'altération de la fonction visuelle est souvent fortuite, souvent associée à la présence de collatérales optociliaires. Les méningiomes peuvent aussi concerner les méninges de la base du crâne. Les schwannomes (souvent improprement appelés « neurinomes » ) peuvent atteindre toutes les paires crâniennes, dont le III, mais aussi des nerfs périphériques, dont des nerfs orbitaires. Des hamartomes combinés de la rétine et de l'épithélium pigmentaire ou des membranes épirétiniennes sont aussi décrits chez un tiers des patients atteints [29]. Sur le plan cutané, les taches café au lait sont rares (< 1 % des patients); les patients présentent souvent des schwannomes nodulaires sous-cutanés, au niveau du tronc dans les formes adultes de NF2. La maladie est principalement connue des ophtalmologistes au travers de sa forme classique, d'évolution lente, dite de type Gardner, généralement révélée par (et parfois limitée à) un schwannome bilatéral du nerf vestibulaire, parfois associé à un méningiome des gaines du nerf optique. Cette forme se rencontre rarement en milieu pédiatrique, car elle survient après l'âge de la puberté.
Chez l'enfant prépubère cependant, la NF2 se présente généralement sous deux formes bien spécifiques [30] : d'une part, la NF2 congénitale, rare; d'autre part, la NF2 de type Wishart, dont la présentation est très différente du classique type Gardner.
La NF2 congénitale se caractérise par :
- –de nombreux schwannomes sous-cutanés en plaques, localisés au niveau des membres et amenés à régresser ultérieurement;
- –des cataractes sous-corticales postérieures discrètes;
- –des anomalies à l'IRM cérébrale : présence de schwannomes bilatéraux et asymptomatiques des nerfs vestibulaires, dont la croissance explosive, en quelques mois, ne surviendra que 10 à 15 ans plus tard; dysplasie corticale initialement asymptomatique mais à risque d'épilepsie ultérieure.
L'expression principalement dermatologique explique que cette forme soit certainement très sous-diagnostiquée.
La NF2 de type Wishart s'exprime par des schwannomes sous-cutanés au niveau des membres; les schwannomes du nerf vestibulaire sont rares tandis que les schwannomes des autres nerfs crâniens (notamment le III, le IV et le VI) sont plus fréquents; une neuropathie périphérique est souvent présente, souvent non expliquée par l'imagerie; les atteintes du système nerveux central (méningiomes et épendymomes) sont fréquentes; les manifestations « oculaires » de la maladie sont parfois révélatrices (cataractes sous-capsulaires souvent peu symptomatiques, hamartomes combinés de la rétine et de l'épithélium pigmentaire présents dans la majorité des cas). L'ensemble de ces atteintes est d'évolution plus rapide que dans les formes de l'adulte.
La sclérose tubéreuse de Bourneville (STB) est une maladie neurocutanée caractérisée par des hamartomes multisystémiques et des manifestations neuropsychiatriques. L'incidence de la maladie est estimée entre 1/6800 et 1/15 000 et la prévalence est de 1/10 000 avec 50 à 84 % des cas sporadiques [31, 32]. C'est une maladie de transmission autosomique dominante à pénétrance variable, causée par l'inactivation, par mutation, des gènes TSC1 ou TSC2 (tuberous sclerosis proteins 1 and 2). Les protéines codées par les gènes TSC1 et TSC2 sont respectivement l'hamartine et la tubérine. Ces protéines forment un complexe qui régule négativement le complexe rapamycine-mTORC1. TORC1 est une kinase qui régule la croissance cellulaire et les processus anaboliques en réponse à une stimulation par un facteur nutritif et la croissance. C'est une maladie hétérogène sur le plan clinique, qui en général est dépistée dans l'enfance mais dont les cas peu sévères sont sous-diagnostiqués [33]. La STB est certaine si le patient réunit : soit deux critères majeurs, soit un critère majeur et deux critères mineurs (encadré 26-3).
4. Sous-partie rédigée par G. Le Meur.
Critères de diagnostic de sclérose tubéreuse de Bourneville
Critères majeurs :
- angiofibrome de la face ;
- fibromes unguéaux ou péri-unguéaux non traumatiques ;
- taches hypomélaniques > 3 ;
- plaque « peau de chagrin » ;
- hamartomes nodulaires rétiniens multiples ;
- tuber cortical ;
- nodule sous-épendymaire ;
- astrocytomes à cellules géantes ;
- rhabdomyome cardiaque unique ou multiple ;
- lymphangioléiomyomatose pulmonaire ;
- angiomyolipome rénal.
Critères mineurs :
- géodes multiples de l’émail dentaire ;
- polypes rectaux hamartomateux ;
- lignes de migration radiaires dans la substance blanche ;
- fibromes gingivaux ;
- hamartomes non rénal ;
- tache rétinienne achrome ;
- lésions cutanées en « confetti » ;
- kystes rénaux multiples.
L'épilepsie est l'un des symptômes les plus invalidants de la STB et est une cause majeure de morbidité et de mortalité chez les personnes touchées : 85 % des enfants atteints présentent une épilepsie précoce (âge moyen de diagnostic 7,5 mois) dont deux tiers seront difficiles à équilibrer par un traitement classique [33]. Il semble qu'il y ait une association avec la survenue de spasmes infantiles et la présence de rhabdomyomes cardiaques ou de kystes rénaux ou de macules cutanées hypopigmentées, ce qui pourrait être un élément clinique prédictif [33]. L'épilepsie serait liée aux tubers (hamartomes) corticaux (fig. 26-12) et au cortex peri-tuber, qui sont des zones génératrices ou propagatrices des activités électriques anarchiques responsables des crises d'épilepsie [34]. L'autre grande complication neurologique de la STB est l'hypertension intracrânienne (HTIC), par hydrocéphalie uni- ou biventriculaire secondaire à l'obstruction du foramen de Monro par un astrocytome sous-épendymaire à cellules géantes (fig. 26-13). Ces astrocytomes sont de croissance lente; l'HTIC n'est pas nécessairement symptomatique avant le stade d'atrophie optique venant conclure un œdème papillaire de stase ancien, en sorte que la surveillance de l'imagerie et du fond d'œil est ici essentielle. Les autres signes neurologiques sont une déficience intellectuelle, des troubles de l'humeur, une anxiété, une plus grande fréquence de dépression, des troubles de l'attention avec une hyperactivité et des troubles du spectre autistique [35].
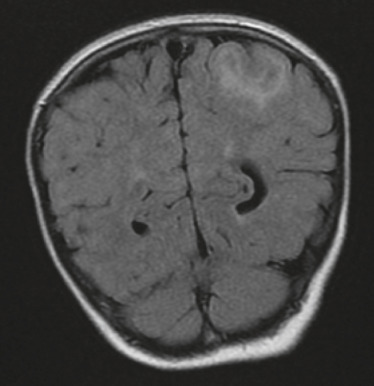
Fig. 26-12 Tubers (hamartomes) corticaux et sous-corticaux dans le cadre d’une sclérose tubéreuse de Bourneville. IRM FLAIR.
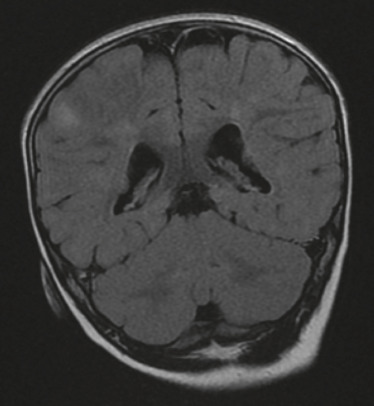
Fig. 26-13 Astrocytomes sous-épendymaires faisant saillie dans la lumière des ventricules latéraux dans le cadre d’une sclérose tubéreuse de Bourneville. IRM FLAIR.
Des macules hypomélaniques en forme de feuille apparaissent au cours de la première année de vie; puis entre 3 et 10 ans ce sont des lésions érythémateuses et papulo-nodulaires (angiofibromes) qui apparaissent notamment au niveau du visage, sur les joues. Des fibromes unguéaux appelés tumeurs de Koenen apparaissent un peu plus tard et sont caractéristiques de la pathologie (fig. 26-14). Au niveau des lombaires et de la tête, des plaques fibreuses dites « plaques en peau de chagrin » peuvent être présentes (fig. 26-15). Ces lésions cutanées, notamment faciales, qui ont des répercussions psychologiques, peuvent être traitées par rapamycine en traitement topique [36].
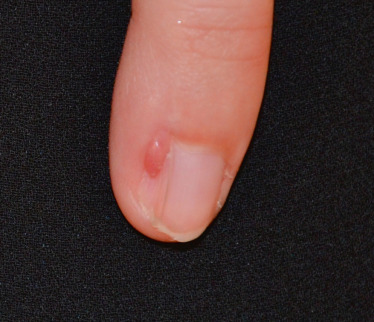
Fig. 26-14 Tumeurs de Koenen dans le cadre d’une sclérose tubéreuse de Bourneville.
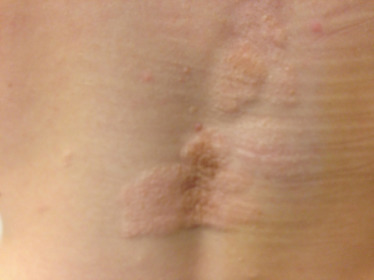
Fig. 26-15 Plaques en peau de chagrin dans le cadre d’une sclérose tubéreuse de Bourneville.
Le rein est le second organe le plus atteint dans la STB, avec des taux d'incidence entre 60 et 75 % . Les deux types de lésions sont les angiomyolipomes et les kystes rénaux. Ces deux lésions peuvent être à l'origine d'une atteinte rénale chronique, voire d'une insuffisance rénale précoce. Ces angiomyolipomes atteignent les reins de manière multiple et bilatérale et peuvent se compliquer d'hémorragie rétropéritonéale, d'hématurie, de saignements rénaux et d'insuffisance rénale [37]. Les complications rénales sont responsables de 30 % des décès survenant dans cette maladie [37].
Cinquante pour cent des personnes atteintes présentent des rhabdomyomes cardiaques généralement identifiés avant la naissance ou au cours de la période néonatale, qui vont spontanément régresser chez une grande majorité des patients au cours de la première année de vie sans donner de symptômes cardiovasculaires [38].
Les tumeurs retrouvées sont des hamartomes astrocytaires rétiniens. Ces lésions sont asymptomatiques mais elles permettent d'apporter des arguments en faveur du diagnostic quand la maladie est suspectée. Cinquante pour cent des patients atteints de STB présentent des hamartomes rétiniens unilatéraux et 25 % d'entre eux présentent des lésions bilatérales [39]. Une classification de ces hamartomes permet de les classer en trois types morphologiques différents. Dans le type 1, les lésions sont circulaires ou de forme ovale, solitaires avec une taille moyenne d'un demi ou d'un diamètre papillaire, situées dans la couche de fibres nerveuses rétiniennes sans signe de calcification (fig. 26-16a). Dans le type 2, plusieurs zones nodulaires calcifiées de taille variable ressemblant à une mÛre sont visualisées au fond d'œil (fig. 26-16b). Dans le type 3, des lésions à la fois du type 1 et du type 2 apparaissent avec un centre calcifié gris blanchâtre avec une périphérie irrégulière semi-translucide (fig. 26-16c) [39]. Ces phacomes rétiniens sont un enchevêtrement de cellules gliales, de foyers de calcification dystrophique et de petits vaisseaux dans la couche superficielle de la rétine. Cela apparaît en coupe OCT (optical coherence tomography) comme un renflement de la couche des ganglionnaires avec plus ou moins une densification vitréenne en regard (fig. 26-17) [10]. Les lésions tubéreuses « jeunes » ne sont pas faciles à identifier en ophtalmoscopie du fait de leur aspect translucide et flou. Elles sont souvent situées à proximité de vaisseaux en donnant l'impression d'une interruption du trajet vasculaire [40]. Avec le temps, il y a une calcification lésionnelle pouvant faire confondre des lésions proches du nerf optique avec des drusen papillaires. Une régression spontanée lésionnelle a également été rapportée [40]. Les autres signes ophtalmologiques sont des taches rétiniennes achromes à l'emporte-pièce ou hyperpigmentées ou des zones de dépigmentation irienne (fig. 26-18) [40].
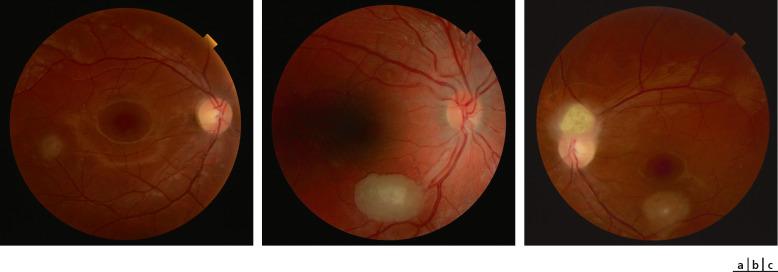
Fig. 26-16 Hamartome astrocytaire rétinien dans le cadre d’une sclérose tubéreuse de Bourneville : types 1 (a), 2 (b) et 3 (c).
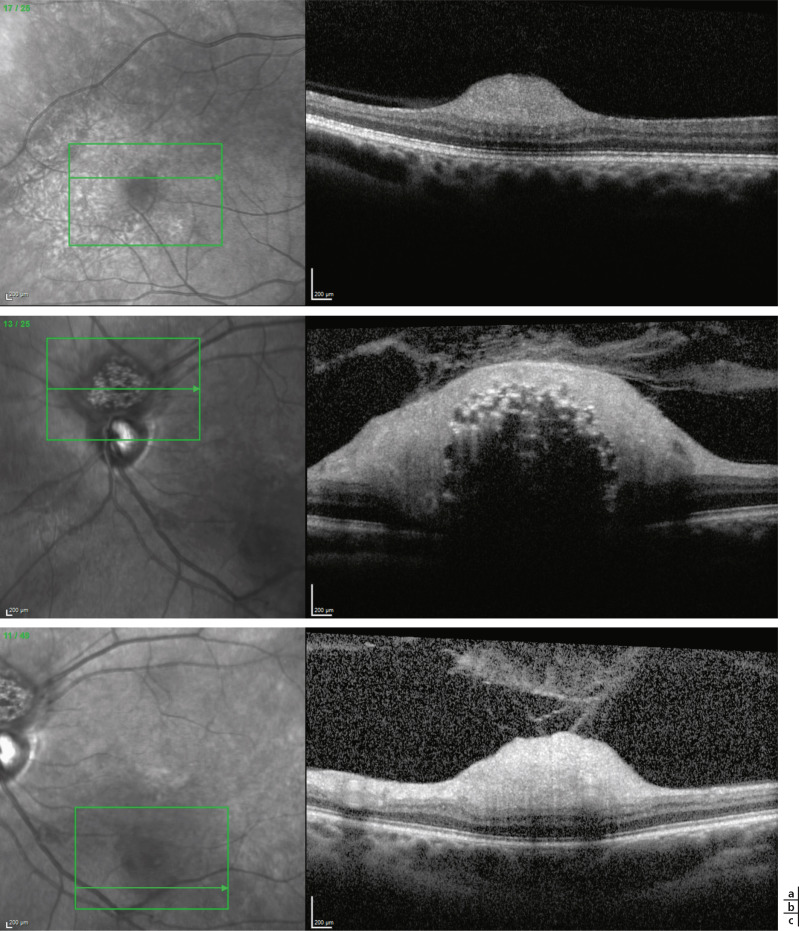
Fig. 26-17 Aspect en spectral-domain optical coherence tomography (SD-OCT) d’hamartomes astrocytaires dans le cadre d’une sclérose tubéreuse de Bourneville.
a. Renflement de la couche des ganglionnaires lié à un hamartome de type 1 (fond d’oeil, même oeil qu’à la fig. 26-16a). b. Hamartome calcifié au niveau de la tête du nerf optique (fond d’oeil, même oeil qu’à la fig. 26-16b). c. Hamartome apparaissant sous la forme d’un renflement de la couche des cellules ganglionnaires avec une densification vitréenne en regard (fond d’oeil, même oeil qu’à la fig. 26-16c).
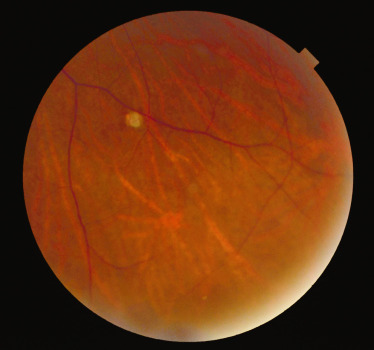
Fig. 26-18 Tache rétinienne achrome à l’emporte-pièce dans le cadre d’une sclérose tubéreuse de Bourneville.
La maladie de von Hippel-Lindau (VHL) est une maladie génétique de prédisposition à des tumeurs bénignes ou malignes qui peuvent survenir dans divers organes : la rétine, les reins, le cerveau, la moelle épinière, les surrénales. C'est une maladie de transmission autosomique dominante à pénétrance élevée (plus de 90 % des patients présentant une mutation du gène VHL développent des symptômes cliniques avant l'âge de 65 ans). Elle est liée à la survenue d'une mutation dans le gène VHL, situé sur le bras court du chromosome 3 [41]. Il s'agit d'un gène suppresseur de tumeur, qui joue un rôle majeur dans la régulation de la transcription et l'expression du vascular endothelial growth factor (VEGF) [42]. L'incidence de la maladie est de 1/36 000 naissances [43]. L'âge d'apparition des premiers signes cliniques peut varier. Le diagnostic est posé si un patient présente deux hémangioblastomes, quelle que soit leur localisation, ou l'association d'un hémangioblastome et d'une autre lésion habituelle [43]. Actuellement dans cette pathologie, il y a une amélioration de l'espérance de vie grâce à un dépistage régulier ciblé au niveau des organes qui peuvent être touchés et à une prise en charge multidisciplinaire.
5. Sous-partie rédigée par G. Le Meur.
La fréquence des hémangioblastomes rétiniens chez les patients atteints de VHL varie selon les études : 58 % pour Maher et al. [44] et 28 % pour Wong et al. [45]. L'atteinte oculaire est fréquemment révélatrice de la maladie. Ces tumeurs rétiniennes, multiples et bilatérales, apparaissent sous la forme de lésions arrondies, saillantes et rosées avec des vaisseaux adjacents tortueux et dilatés. En général, l'atteinte est en périphérie rétinienne, plus fréquemment en temporal, mais une atteinte papillaire est possible (8 % ) et rarement du pôle postérieur (1 % ). Une exsudation peut apparaître si la lésion évolue (fig. 26-19). Le signe clinique est une baisse d'acuité visuelle, mais actuellement les lésions sont découvertes à un stade asymptomatique du fait d'une surveillance systématique du fond d'œil chez les patients atteints. Les complications sont le décollement de rétine, la cataracte ou le glaucome. Un traitement par laser si la lésion n'est pas trop surélevée ou par thermothérapie transpupillaire est recommandé, voire une cryothérapie si la lésion est trop épaisse. La réalisation d'une chirurgie vitréorétinienne avec ablation de l'hémangioblastome a récemment été décrite en cas de décollement de rétine associé à un traitement laser infructueux; l'utilisation bénéfique de laser photodynamic therapy (PDT) a aussi été rapportée [46, 47]. Les injections intravitréennes d'anti-VEGF ne semblent pas avoir d'effets bénéfiques sur l'exsudation dans ces cas [48]. La réalisation d'un premier fond d'œil est recommandée dès l'âge de 5 ans avec un contrôle annuel.
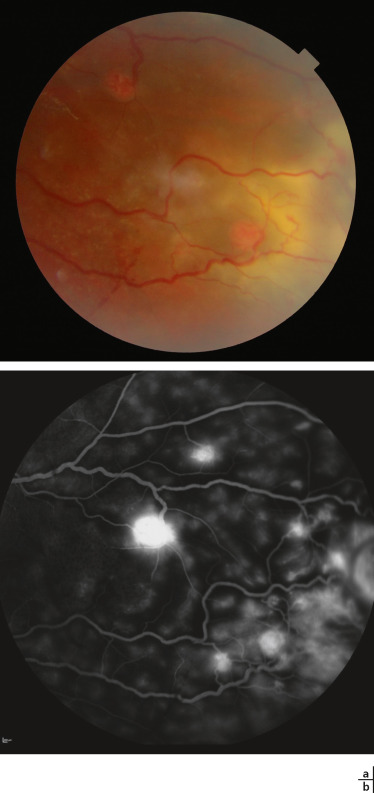
Fig. 26-19 Hémangioblastome rétinien dans le cadre d’une maladie de von Hippel-Lindau.
a. Photographie du fond d’oeil : les tumeurs rétiniennes apparaissent sous la forme de lésions arrondies, saillantes et rosées avec des vaisseaux adjacents tortueux et dilatés. b. Angiographie à la fluorescéine montrant une dilatation vasculaire et une exsudation au niveau d’hémangioblastomes rétiniens qui apparaissent hyperfluorescents avec diffusion de fluorescéine.
Le cerveau est l'organe le plus touché par les tumeurs, 70 % des patients ayant cette maladie développent des hémangioblastomes du système nerveux central dans la seconde décade. Les atteintes neurologiques par compression font souvent poser le diagnostic. Ces hémangioblastomes sont le plus souvent multiples et souvent situés dans le cervelet (52 % ), la moelle épinière (44 % ) et le tronc cérébral (18 % ) [49]. L'évolution tumorale n'est pas prédictible et est marquée par une alternance de phase de stabilité et de phase de croissance tumorale rapide. Une imagerie cérébrale, dès l'âge de 10 ans, doit être réalisée chez les patients atteints.
La tumeur la plus fréquente dans le VHL est le carcinome rénal à cellules claires. D'ailleurs, la maladie de VHL est la cause la plus fréquente de cancer du rein familial. L'atteinte rénale est présente dans 30 à 70 % des cas mais la différence par rapport à une atteinte sporadique est l'âge de survenue plus précoce, vers 40 ans, de cette tumeur. C'est la cause de décès chez les patients atteints de VHL. Un aspect de polykystose rénale est souvent associé.
Le pancréas est fréquemment atteint dans la maladie de VHL sous la forme soit de kystes pancréatiques simples, soit de cystadénomes ( fig. 26-20), soit de tumeurs neuro-endocrines et plus rarement d'adénocarcinomes.
Parmi les autres atteintes tumorales possibles, des cystadénomes de l'épididyme ou du ligament large peuvent être responsables de troubles de la fertilité. Un phéochromocytome ( fig. 26-21), souvent bilatéral, est associé dans 11 à 20 % des cas de VHL, responsable d'hypertension artérielle paroxystique ou continue. Des tumeurs du sac endolymphatique avec une perte de l'audition dans 10 % des cas sont rapportées. Les manifestations rares peuvent être des kystes et angiomes dans le foie, la rate, les poumons ou les os, qui ne sont généralement diagnostiqués que fortuitement.
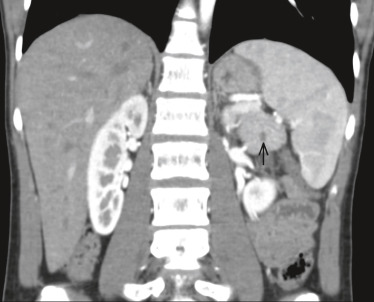
Fig. 26-20 Cystadénome pancréatique (flèche) dans le cadre d’une maladie de von Hippel-Lindau.
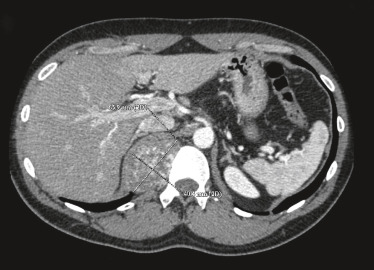
Fig. 26-21 Phéochromocytome dans le cadre d’une maladie de von Hippel-Lindau.
Il s'agit d'un ensemble d'affections à la physiopathologie imparfaitement comprise, qui se caractérisent par des malformations vasculaires cutanées, cérébrales et/ou oculaires et qui, selon les territoires affectés, sont désignées par les noms variés d'auteurs les ayant décrites.
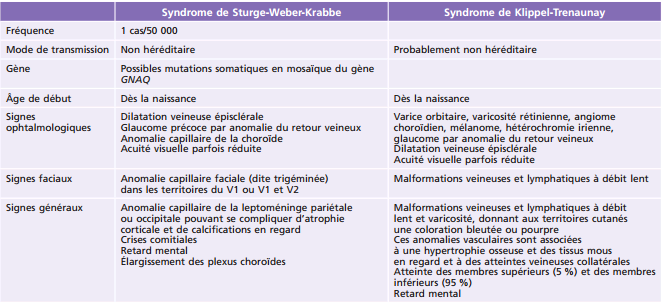
Les caractéristiques des syndromes de Sturge-Weber-Krabbe et de Klippel-Trenaunay sont décrites dans le tableau 26-2 [50 - 52]. Le terme « angiome plan » , qui désignait classiquement les anomalies cutanées communes à ces syndromes (aussi appelées tache lie-de-vin), est désormais évité (ainsi que son corollaire), car un angiome est une lésion proliférative, tandis qu'ici l'anomalie est de type malformatif, stable, et est donc plus justement désignée par le terme de malformation capillaire. L'atteinte peut concerner variablement la peau, les yeux et le cerveau. Le terme syndrome de Sturge-Weber-Krabbe désigne classiquement une triade atteinte cutanée, cérébrale et ophtalmologique. On parle souvent de forme complète dans ces cas. L'usage désigne désormais aussi les formes incomplètes (atteinte cutanée ou cérébrale) sous le même nom. Certains ont proposé une classification en trois types : type I – atteinte cutanée et cérébrale, avec ou sans atteinte oculaire; type II – atteinte cutanée avec ou sans atteinte oculaire mais sans atteinte cérébrale; type III – atteinte cérébrale sans atteinte cutanée ni oculaire [53]. Sur le plan pratique, il semble que le principal facteur de risque d'atteinte oculaire soit l'atteinte de la paupière supérieure, justifiant d'une surveillance spécifique et à vie les enfants présentant une malformation capillaire dans cette localisation.
6. Sous-partie rédigée par C. Orssaud, M. Robert.
On désigne parfois sous le nom de syndrome de Shapiro-Shulman l'association : malformation capillaire impliquant le territoire facial inférieur et cervical supérieur, macrocrânie, hypertension veineuse intracrânienne et anomalies du retour veineux. Il s'agit probablement de l'expression cliniquement distincte d'un mécanisme identique à celui du syndrome de Sturge-Weber-Krabbe.
Le syndrome de Bonnet-Dechaume-Blanc est une pathologie sporadique au cours de laquelle il existe une malformation artérioveineuse congénitale rétinienne et cérébrale unilatérale. Plusieurs termes ont été utilisés pour qualifier cette communication artérioveineuse congénitale anormale qui est plus ou moins sévère [54]. Il est classique de distinguer quatre groupes de gravité différente. Les premier et deuxième groupes diffèrent des deux autres par la présence ou l'absence d'anomalies vasculaires dans les territoires cutanés faciaux homolatéraux aux lésions oculocérébrales, notamment au niveau palpébral. Le troisième groupe correspond à une atteinte oculaire isolée. Les patients du dernier groupe ont une atteinte oculocérébrale mais sans anomalie vasculaire décelable en neuroradiologie [54].
Ce syndrome qui se révèle précocement, dès l'âge de 4 ans, est généralement responsable d'une altération de l'acuité visuelle plus ou moins marquée allant jusqu'à une profonde malvoyance ou une cécité. Mais l'altération de la fonction visuelle peut être due à l'atteinte des voies visuelles intracérébrales par l'anomalie artérioveineuse. Les anomalies vasculaires intéressent la totalité de la rétine ou restent localisées à un secteur, notamment en périphérie. Elles se compliquent, chez certains patients, d'hémorragie du vitré ou d'hémorragie maculaire [55, 56]. Le pronostic visuel est meilleur dans le troisième groupe qui s'accompagne d'une atteinte rétinienne plus volontiers partielle [54]. Des troubles oculomoteurs ont été rapportés, conséquence d'accidents vasculaires cérébraux secondaires à la présence de la malformation et pouvant s'exprimer dans l'enfance.
Ces atteintes neurologiques s'observent dans les trois groupes comportant des malformations artérioveineuses cérébrales identifiées ou non. Il a été décrit, outre des céphalées et la perception d'un souffle, des atteintes des nerfs crâniens, des hémiparésies et des hémorragies méningées. De tels accidents ont été rapportés chez des enfants dès l'âge de 6 ans [54]. Les anomalies vasculaires cérébrales ont tendance à régresser avec l'âge et à se calcifier. Une embolisation de l'anomalie cérébrale peut être tentée [56].
L'ataxie-télangiectasie (AT) de Denise Louis-Bar est une pathologie récessive autosomique associée à des mutations du gène ATM codant une protéine kinase impliquée dans la régulation du cycle cellulaire et la réparation des cassures des brins d'acide désoxyribonucléique (ADN) [57]. Son rôle est donc proche de celui des gènes suppresseurs de tumeur. C'est pourquoi il existe, au cours de l'AT, une sensibilité de la chromatine aux radiations ionisantes associée à des cassures de l'ADN qui peut être recherchée sur des lymphocytes et constituer un test diagnostique.
L'apraxie oculomotrice apparaît précocement. Elle réalise un défaut d'initiation des saccades horizontales et verticales, ce qui la différencie de l'apraxie observée dans le syndrome de Cogan. D'autres troubles oculomoteurs peuvent être retrouvés à type d'altération de la poursuite, de nystagmus et de strabisme; ils sont présents chez plus de 30 % des patients. Les classiques télangiectasies conjonctivales apparaissent entre 3 et 5 ans chez la plupart des enfants homozygotes pour les mutations de ce gène. Il n'est pas rare qu'elles concernent également la face.
L'atteinte neurologique est précoce avec apparition d'une ataxie cérébelleuse causée par la dégénérescence des cellules de Purkinje. Elle est d'autant plus invalidante qu'il s'y associe des mouvements choréo-athétosiques, une neuropathie périphérique, aboutissant à une abolition des réflexes tendineux et une fonte musculaire, et un syndrome extrapyramidal. Un retard mental est décrit mais très inconstant. Ces enfants contractent volontiers des infections des voies aériennes supérieures ou des bronches avec formation de bronchectasies en raison d'un déficit immunitaire. La synthèse des immunoglobulines A (IgA) et des IgG, et à un degré moindre des IgM et des IgE, est diminuée.
Une surveillance des patients homozygotes s'impose toute la vie en raison du risque d'apparition d'une leucémie lymphoïde de type T, d'un lymphome malin de type B ou d'une tumeur maligne solide. Le risque de survenue d'une tumeur maligne, notamment au niveau du sein chez la femme, est également rapporté chez les patients hétérozygotes.
7. Sous-partie rédigée par C. Orssaud.
M. Robert
L'ichtyose désigne une peau à l'aspect sec et craquelé, typiquement en écaille de poisson (ichtus), résultant d'une desquamation cutanée continue. La majorité des ichtyoses sont dystrophiques, donc génétiquement déterminées. Leur mode de révélation, cependant, va des formes congénitales sévérissimes à des manifestations discrètes sans plainte fonctionnelle chez un jeune adulte. Les ichtyoses sévères requièrent chez le nourrisson une prise en charge spécialisée où l'ophtalmopédiatre joue un rôle crucial dans la prévention des complications cornéennes et l'éducation des parents. Parmi les ichtyoses non syndromiques, les ichtyoses liées à l'X comportent des opacités cornéennes stromales ou descemétiques dans 50 % des cas environ chez les garçons atteints et leurs mères.
La plupart des ichtyoses syndromiques affectant les yeux appartiennent au groupe des syndromes neuro-ichtyosiques. Plusieurs d'entre eux (notamment les syndromes de Sjögren-Larsson, de Refsum et la maladie de Gaucher de type II) sont des maladies métaboliques et sont détaillés également dans le chapitre 27.7. Les caractéristiques des ichtyoses syndromiques comportant des manifestations ophtalmologiques sont indiquées dans le tableau 26-3 [58].
Tableau 26-3 – Caractéristiques des syndromes ichtyosiques affectant les yeux.
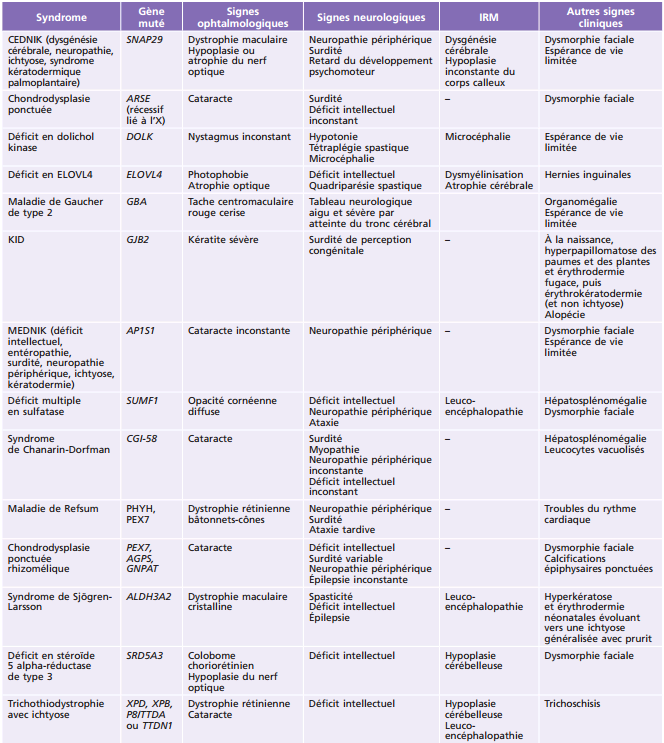
CEDNIK : cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma ; KID : keratite-ichthyosis-deafness ; IRM : imagerie par résonance magnétique ; MEDNIK : mental retardation, enteropathy, deafness, peripheral neuropathy, ichthyosis, and keratoderma.
C. Orssaud
Le pseudoxanthome élastique (PXE) est une pathologie héréditaire du tissu élastique, transmise selon un mode autosomique récessif et liée à une mutation du gène ABCC6, localisé en 16p13. Ce gène code une protéine transmembranaire de la famille C des « protéines à cassettes liant l'adénosine triphosphate (ATP) » dont la fonction exacte est inconnue [59, 60]. La prévalence du PXE est de 1/50 000 à 1/100 000 mais reste sans doute sous-estimée. Il toucherait plus volontiers les femmes, avec une sex-ratio de 2: 1.
Au cours du PXE, les atteintes cliniques intéressent la peau, l'œil et le système vasculaire dans son ensemble et sont dues à une lente calcification des fibres élastiques de ces tissus. Les anomalies cutanées apparaissent dans l'enfance ou l'adolescence et sont d'abord localisées au niveau du cou (donnant l'aspect en cou de poulet) et à la partie postérieure des grosses articulations. Les complications vasculaires sont habituellement d'apparition plus tardive, à l'âge adulte et se manifestent par la survenue de rétrécissements et occlusions des artères de moyen calibre aboutissant à une hypertension artérielle, une claudication des membres, des accidents vasculaires transitoires ou des infarctus myocardiques [61, 62].
Les manifestations ophtalmologiques intéressent la choriorétine et associent dans le temps un aspect en peau d'orange, puis des stries angioïdes qui peuvent se compliquer de néovascularisation choriorétinienne et de cicatrices maculaires. Ces anomalies ne sont pas nécessairement retrouvées chez l'enfant [63, 64]. Les manifestations et complications ophtalmologiques sont favorisées par les traumatismes, parfois minimes, y compris chez l'enfant [65].
Le xeroderma pigmentosum (XP) est une pathologie rare hétérogène, dont il existe huit types dus à la présence de mutations dans huit gènes différents. Ces gènes codent des protéines impliquées dans la réparation de l'ADN. La forme « variant » du XP n'intéresse pas l'ophtalmopédiatrie. La perte de fonction de ces protéines est responsable d'une sensibilité accrue à la lumière et aux rayons ultraviolets (UV), ce rayonnement étant délétère pour l'ADN. Cliniquement, le XP est caractérisé par la survenue de lentigines avant l'âge de 2 ans et de cancers au niveau des zones cutanées et des muqueuses exposées à la lumière. Les cancers cutanés basocellulaires apparaissent généralement avant l'âge de 10 ans et sont localisés dans 80 % des cas au niveau de la tête et du cou. En revanche, il est fréquent que les cancers muqueux évoluent vers un cancer spinocellulaire. Des mélanomes se développent généralement après l'âge de 20 ans. Cependant, leur présence chez l'enfant n'est pas exceptionnelle. Leur répartition est différente de celles des cancers basocellulaires. Il faut noter que l'anomalie de réparation de l'ADN affecte également les neurones qui présentent une sensibilité accrue au métabolisme oxydatif. C'est pourquoi le XP se complique de tumeurs cérébrales avant l'âge de 20 ans et d'une dégénérescence neurologique progressive à l'âge adulte.
Les atteintes ophtalmologiques apparaissent précocement. Elles sont retrouvées dans 50 à 80 % des cas, intéressent les annexes oculaires et le segment antérieur de l'œil. Au niveau palpébral, il est volontiers retrouvé, outre les lentigines des paupières, des plages d'atrophie ainsi que des tumeurs bénignes (papillomes) ou malignes. Il s'agit de tumeurs de type basocellulaire plus que spinocellulaire. L'atteinte conjonctivale est plus rare, sous forme de ptérygions ou pseudo-ptérygions, de carcinomes basocellulaires ou surtout épidermoïdes. Ceux-ci ont tendance à s'étendre à la cornée ou à métastaser. La présence d'un syndrome sec compliqué de kératite d'exposition est volontiers rapportée et participe à la survenue des altérations cornéennes. Celles-ci semblent secondaires à la toxicité directe des rayons UV mais leur chronologie est mal déterminée. Ces altérations cornéennes intéressent l'épithélium, la membrane de Bowman et le stroma avec développement d'une kératite bulleuse, d'une néovascularisation s'étendant dans le stroma, d'un pannus, mais aussi d'ulcérations et perforations. Les altérations concernent également la membrane de Descemet qui est épaissie et les cellules endothéliales dont la perte précoce et accélérée a bien été démontrée. Le stade ultime est marqué par une opacification cornéenne plus ou moins étendue. Les manifestations ophtalmologiques et générales observées en fonction du type de XP sont présentées dans le tableau 26-4.
La prise en charge ophtalmologique repose sur l'ablation des tumeurs suivie de reconstruction tissulaire. La greffe de membrane amniotique permet de traiter les altérations épithéliales les plus sévères en soulageant le patient. Mais, les opacifications cornéennes et les altérations endothéliales peuvent nécessiter la réalisation de kératoplasties, parfois à un âge précoce.
Tableau 26-4 – Manifestations ophtalmologiques et générales observées en fonction du type de xeroderma pigmentosum.
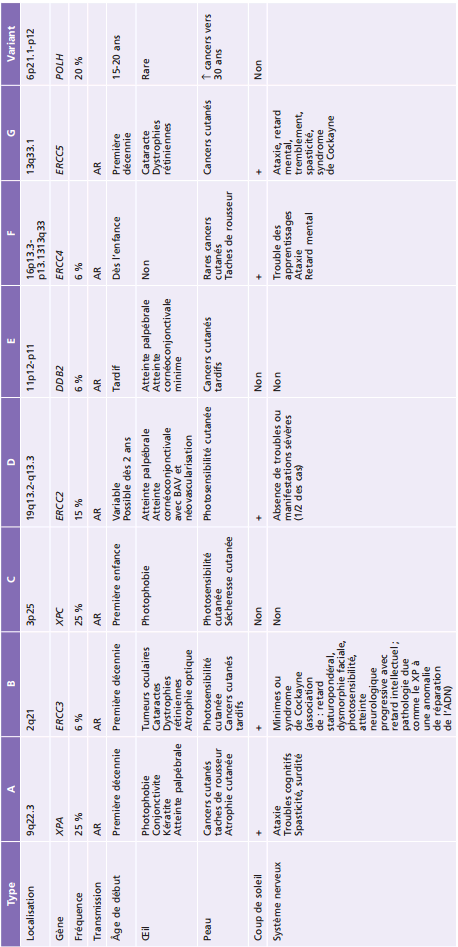
AR : autosomique récessive ; BAV : baisse de l’acuité visuelle.
Le xanthogranulome juvénile (XGJ) est une pathologie rare, d'étiologie encore imprécise, entrant dans le groupe des histiocytoses non langheransiennes [66 - 68]. Il est développé à partir de cellules dendritiques du derme. Cette origine explique pourquoi le XGJ se manifeste par la survenue de tumeurs bénignes, uniques ou multiples au niveau cutané ou muqueux. Néanmoins, il existe des localisations extracutanées du XGJ au niveau hépatique, cardiaque, splénique, rénal, du système nerveux central et surtout oculaire. Cette atteinte oculaire intéresse essentiellement l'uvée. Mais d'autres structures oculaires et extra-oculaires peuvent être intéressées, telles que la cornée, la conjonctive ou les paupières [68]. Ces xanthogranulomes apparaissent dans l'enfance ou à l'adolescence. Le diagnostic est histologique. Il retrouve un infiltrat constitué d'histiocytes spumeux, de lymphocytes et de cellules géantes de Touton, cellules plurinucléées à cytoplasme éosinophile. Cet aspect permet d'éliminer d'autres pathologies histiocytaires ou lymphoprolifératives. Les atteintes cutanées ont tendance à régresser spontanément. En revanche, au niveau oculaire, une corticothérapie locale est indispensable pour faire régresser les lésions iriennes afin d'éviter les complications parfois révélatrices, aux premiers rangs desquelles il faut citer l'hyphéma récidivant et l'hypertonie oculaire. Les atteintes cornéolimbique ou palpébrale doivent être retirées chirurgicalement car elles n'ont pas tendance à régresser spontanément (voir chapitre 10).
Le syndrome de Stevens-Johnson (SSJ) est une pathologie inflammatoire aiguë, généralement secondaire à la prise de médicaments, qui touche la peau et les muqueuses, mais qui intéresse moins de 10 % de la surface cutanée. La forme la plus sévère, touchant plus de 30 % de la surface cutanée, est appelée syndrome de Lyell (SLy) ou épidermolyse bulleuse. Elle ne représente que 20 % des cas [69]. Cet ensemble pathologique n'est pas exceptionnel, avec une prévalence de 2 cas par million d'habitant [70]. L'atteinte de la sphère oculaire n'est pas exceptionnelle et concerne entre 75 et 100 % des enfants atteints de SSJ ou SLy. Elle apparaît en même temps que l'atteinte cutanée, mais peut être retardée de quelques jours. Les atteintes liées aux SSJ/SLy intéressent précocement la conjonctive, l'épithélium cornéen et le tarse. L'atteinte de la sphère oculaire débute par une inflammation de l'ensemble de la surface oculaire et des paupières à laquelle s'associent volontiers des pseudo-membranes. Des érosions tarsales, conjonctivales ou de l'épithélium et du stroma cornéen apparaissent dans un tiers des cas environ dans les jours suivants [69]. Celles-ci sont plus ou moins étendues en surface et en profondeur. Dans les formes les plus graves et sévères, sont observées des ulcérations ou des perforations cornéennes. Contrairement aux lésions cutanées, qui guérissent sans séquelle notable, l'atteinte ophtalmologique peut se compliquer. Une baisse d'acuité visuelle définitive doit être prévenue dans les formes les plus sévères et notamment chez les patients les plus jeunes [71]. Elle reste heureusement rare dans une série pédiatrique récente puisqu'elle ne concerne que 8 % des enfants [69]. Mais la persistance d'un inconfort plusieurs mois après l'épisode aigu reste très fréquente, y compris lors d'atteintes oculaires a priori peu sévères.
En effet, les atteintes cornéennes font toute la gravité visuelle du SSJ, puisqu'elles cicatrisent parfois au prix d'une opacification et néovascularisation cornéenne. Il faut insister sur le risque d'amblyopie définitive liée aux anomalies de transparence cornéenne chez le jeune enfant en l'absence de restauration rapide de la transparence cornéenne. Au niveau conjonctival, les lésions du SSJ sont responsables de kératinisation du bord libre palpébral et de dysfonction des glandes de Meibomius, de symblépharon et ankyloblépharon entraînant parfois une sténose du point lacrymal. Ces complications aboutissent à des troubles plus ou moins marqués de la statique palpébrale, avec entropion et distichiasis, et à une sécheresse oculaire, deux éléments qui majorent les altérations de la surface cornéenne à long terme et la sensation d'inconfort.
Le traitement n'est pas codifié. Il repose sur la prescription systématique de collyres mouillants. Une corticothérapie topique peut être utile à la phase aiguë dans les formes sévères [72]. Enfin, la greffe de membrane amniotique, réalisée précocement, permet de diminuer le risque d'opacification cornéenne résiduelle dans les formes graves [73].
[1] Abramowicz A, Gos M Neurofibromin in neurofibromatosis type 1 – mutations in NF1gene as a cause of disease Developmental Period Medicine ( 2014 ) : 18: 297-306
[2] Rauen KA, Huson SM, Burkitt-Wright E Recent developments in neurofibromatoses and RASopathies : management, diagnosis and current and future therapeutic avenues Am J Med Genet A ( 2015 ) : 167A: 1-10
[3] Upadhyaya M, Huson SM, Davies M An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT) : evidence of a clinically significant NF1 genotype-phenotype correlation Am J Hum Genet ( 2007 ) : 80: 140-151
[4] Pasmant E, Sabbagh A, Spurlock G NF1 microdeletions in neurofibromatosis type 1 : from genotype to phenotype Hum Mutat ( 2010 ) : 31: E1506-E1518
[5] Venturin M, Guarnieri P, Natacci F Mental retardation and cardiovascular malformations in NF1 microdeleted patients point to candidate genes in 17q11.2 J Med Genet ( 2004 ) : 41: 35-41
[6] Boyd KP, Korf BR, Theos A Neurofibromatosis type 1 J Am Acad Dermatol ( 2009 ) : 61: 1-14 quiz 5-6
[7] Listernick R, Charrow J, Greenwald M, Mets M Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study J Pediatr ( 1994 ) : 125: 63-66
[8] Blazo MA, Lewis RA, Chintagumpala MM Outcomes of systematic screening for optic pathway tumors in children with Neurofibromatosis Type 1 Am J Med Genet A ( 2004 ) : 127A: 224-229
[9] Blanchard G, Lafforgue MP, Lion-Francois L Systematic MRI in NF1 children under six years of age for the diagnosis of optic pathway gliomas. Study and outcome of a French cohort Eur J Paediatr Neurol ( 2016 ) : 20: 275-281
[10] Fisher MJ, Loguidice M, Gutmann DH Visual outcomes in children with neurofibromatosis type 1-associated optic pathway glioma following chemotherapy : a multicenter retrospective analysis Neuro Oncol ( 2012 ) : 14: 790-797
[11] Avery RA, Dombi E, Hutcheson KA Visual outcomes in children with neurofibromatosis type 1 and orbitotemporal plexiform neurofibromas Am J Ophthalmol ( 2013 ) : 155: 1089-1094 e1
[12] Viola F, Villani E, Natacci F Choroidal abnormalities detected by near-infrared reflectance imaging as a new diagnostic criterion for neurofibromatosis 1 Ophthalmology ( 2012 ) : 119: 369-375
[13] Muci-Mendoza R, Ramella M, Fuenmayor-Rivera D Corkscrew retinal vessels in neurofibromatosis type 1 : report of 12 cases Br J Ophthalmol ( 2002 ) : 86: 282-284
[14] Stevenson DA, Yan J, He Y Multiple increased osteoclast functions in individuals with neurofibromatosis type 1 Am J Med Genet A ( 2011 ) : 155A: 1050-1059
[15] Morcaldi G, Clementi M, Lama G Evaluation of tibial osteopathy occurrence in neurofibromatosis type 1 Italian patients Am J Med Genet A ( 2013 ) : 161A: 927-934
[16] Heerva E, Koffert A, Jokinen E A controlled register-based study of 460 neurofibromatosis 1 patients : increased fracture risk in children and adults over 41 years of age J Bone Miner Res ( 2012 ) : 27: 2333-2337
[17] Jaremko JL, MacMahon PJ, Torriani M Whole-body MRI in neurofibromatosis : incidental findings and prevalence of scoliosis Skeletal Radiol ( 2012 ) : 41: 917-923
[18] Hirbe AC, Gutmann DH Neurofibromatosis type 1 : a multidisciplinary approach to care Lancet Neurology ( 2014 ) : 13: 834-843
[19] Arrington DK, Danehy AR, Peleggi A Calvarial defects and skeletal dysplasia in patients with neurofibromatosis Type 1 J Neurosurg Pediatr ( 2013 ) : 11: 410-416
[20] Karvonen M, Saari A, Hannila ML Elevated head circumference-to-height ratio is an early and frequent feature in children with neurofibromatosis type 1 Hormone Research in Paediatrics ( 2013 ) : 79: 97-102
[21] Loitfelder M, Huijbregts SC, Veer IM Functional connectivity changes and executive and social problems in neurofibromatosis type I Brain Connectivity ( 2015 ) : 5: 312-320
[22] Descheemaeker MJ, Ghesquiere P, Symons H Behavioural, academic and neuropsychological profile of normally gifted neurofibromatosis type 1 children J Intellect Disabil Res ( 2005 ) : 49: 33-46
[23] Hyman SL, Shores A, North KN The nature and frequency of cognitive deficits in children with neurofibromatosis type 1 Neurology ( 2005 ) : 65: 1037-1044
[24] Miguel CS, Chaim-Avancini TM, Silva MA, Louza MR Neurofibromatosis type 1 and attention deficit hyperactivity disorder : a case study and literature review Neuropsychiatric Disease and Treatment ( 2015 ) : 11: 815-821
[25] Roy A, Roulin JL, Charbonnier V Executive dysfunction in children with neuro fibromatosis type 1 : a study of action planning J Int Neuropsychol Soc ( 2010 ) : 16: 1056-1063
[26] Hachon C, Iannuzzi S, Chaix Y Behavioural and cognitive phenotypes in children with neurofibromatosis type 1 (NF1) : the link with the neurobiological level Brain Dev ( 2011 ) : 33: 52-61
[27] Hua C, Zehou O, Ducassou S Sirolimus improves pain in NF1 patients with severe plexiform neurofibromas Pediatrics ( 2014 ) : 133: e1792-e1797
[28] Evans DG Neurofibromatosis type 2 (NF2) : a clinical and molecular review Orphanet J Rare Dis ( 2009 ) : 4: 16 p
[29] Feucht M, Griffiths B, Niemuller I Neurofibromatosis 2 leads to higher incidence of strabismological and neuro-ophthalmological disorders Acta Ophthalmol ( 2008 ) : 86: 882-886
[30] Ruggieri M, Pratico AD, Evans DG Diagnosis, management, and new therapeutic options in childhood neurofibromatosis type 2 and related forms Semin Pediatr Neurol ( 2015 ) : 22: 240-258
[31] Sahin M, Henske EP, Manning BD Advances and future directions for tuberous sclerosis complex research : recommendations from the 2015 Strategic Planning Conference Pediatr Neurol ( 2016 ) : 60: 1-12
[32] Wiederholt WC, Gomez MR, Kurland LT Incidence and prevalence of tuberous sclerosis in Rochester, Minnesota, 1950 through 1982 Neurology ( 1985 ) : 35: 600-603
[33] Jeong A, Wong M Systemic disease manifestations associated with epilepsy in tuberous sclerosis complex Epilepsia ( 2016 ) : 57: 1443-1449
[34] Kannan L, Vogrin S, Bailey C Centre of epileptogenic tubers generate and propagate seizures in tuberous sclerosis Brain ( 2016 ) : 139: 2653-2667
[35] Jeste SS, Varcin KJ, Hellemann GS Symptom profiles of autism spectrum disorder in tuberous sclerosis complex Neurology ( 2016 ) : 87: 766-772
[36] Bougueon G, Lagarce F, Martin L Formulation and characterization of a 0.1 % rapamycin cream for the treatment of tuberous sclerosis complex-related angiofibromas Int J Pharm ( 2016 ) : 509: 279-284
[37] Bissler J, Cappell K, Charles H Long-term clinical morbidity in patients with renal angiomyolipoma associated with tuberous sclerosis complex Urology ( 2016 ) : 95: 80-87
[38] Jozwiak S, Kotulska K, Kasprzyk-Obara J Clinical and genotype studies of cardiac tumors in 154 patients with tuberous sclerosis complex Pediatrics ( 2006 ) : 118: e1146-e1151
[39] Mennel S, Meyer CH, Eggarter F, Peter S Autofluorescence and angiographic findings of retinal astrocytic hamartomas in tuberous sclerosis Ophthalmologica ( 2005 ) : 219: 350-356
[40] Abdolrahimzadeh S, Plateroti AM, Recupero SM, Lambiase A An update on the ophthalmologic features in the phakomatoses J Ophthalmol ( 2016 ) : 2016: 3043026 p
[41] Crossey PA, Foster K, Richards FM Molecular genetic investigations of the mechanism of tumourigenesis in von Hippel-Lindau disease : analysis of allele loss in VHL tumours Human Genetics ( 1994 ) : 93: 53-58
[42] Haddad NM, Cavallerano JD, Silva PS Von hippel-lindau disease : a genetic and clinical review Seminars in Ophthalmology ( 2013 ) : 28: 377-386
[43] Ben-Skowronek I, Kozaczuk S Von Hippel-Lindau Syndrome Hormone Research in Paediatrics ( 2015 ) : 84: 145-152
[44] Maher ER, Yates JR, Harries R Clinical features and natural history of von Hippel-Lindau disease Q J Med ( 1990 ) : 77: 1151-1163
[45] Wong M, Chu YH, Tan HL Clinical and molecular characteristics of East Asian patients with von Hippel-Lindau syndrome Chinese Journal of Cancer ( 2016 ) : 35: 79 p
[46] Avci R, Yilmaz S, Inan UU Vitreoretinal surgery for patients with severe exudative and proliferative manifestations of retinal capillary hemangioblastoma because of Von Hippel-Lindau Disease Retina ( 2016 )
[47] Bhattacharjee H, Deka H, Deka S Verteporfin photodynamic therapy of retinal capillary hemangioblastoma in von Hippel-Lindau disease Indian J Ophthalmol ( 2010 ) : 58: 73-75
[48] Wong WT, Liang KJ, Hammel K Intravitreal ranibizumab therapy for retinal capillary hemangioblastoma related to von Hippel-Lindau disease Ophthalmology ( 2008 ) : 115: 1957-1964
[49] Filling-Katz MR, Choyke PL, Oldfield E Central nervous system involvement in Von Hippel-Lindau disease Neurology ( 1991 ) : 41: 41-46
[50] Thomas-Sohl KA, Vaslow DF, Aria BL Sturge-Weber syndrome : a review Pediatr Neurol ( 2004 ) : 30: 303-310
[51] Parsa CF Focal venous hypertension as a pathophysiologic mechanism for tissue hypertrophy, port-wine stains, the Sturge-Weber syndrome, and related disorders : proof of concept with novel hypothesis for underlying etiological cause (an American Ophthalmological Society thesis) Trans Am Ophthalmol Soc ( 2013 ) : 111: 180-215
[52] Cohen MM Klippel-Trenaunay syndrome Am J Med Genet ( 2000 ) : 93: 171-175
[53] Roach E, Bodensteiner J Neurologic manifestations of Sturge-Weber syndrome Sturge-Weber Syndrome: Sturge-Weber Foundation ( 1999 ) 27-38
[54] Schmidt D, Pache M, Schumacher M The congenital unilateral retinocephalic vascular malformation syndrome (bonnet-dechaume-blanc syndrome or wyburn-mason syndrome) : review of the literature Surv Ophthalmol ( 2008 ) : 53: 227-249
[55] Saleh M, Gaucher D, Sauer A Analyse en tomographie par cohérence optique spectrale d'une malformation artério-veineuse rétinienne (syndrome de Wyburn-Mason) J Fr Ophtalmol ( 2009 ) : 32: 779-780
[56] Onder HI, Alisan S, Tunc M Serous retinal detachment and cystoid macular edema in a patient with Wyburn-Mason syndrome Semin Ophthalmol ( 2013 )
[57] Andegeko Y, Moyal L, Mittelman L Nuclear retention of ATM at sites of DNA double strand breaks J Biol Chem ( 2001 ) : 276: 38224-38230
[58] Rizzo WB, Malone Jenkens S, Boucher P Recognition and diagnosis of neuroichthyotic syndromes Semin Neurol ( 2012 ) : 32: 75-84
[59] Ringpfeil F, Lebwohl MG, Christiano AM, Uitto J Pseudoxanthoma elasticum : mutations in the MRP6 gene encoding a transmembrane ATP-binding cassette (ABC) transporter Proc Natl Acad Sci U S A ( 2000 ) : 97: 6001-6006
[60] Germain DP Pseudoxanthoma elasticum : evidence for the existence of a pseudogene highly homologous to the ABCC6 gene J Medical Genet ( 2001 ) : 38: 457-461
[61] Germain DP, Boutouyrie P, Laloux B, Laurent S Arterial remodeling and stiffness in patients with pseudoxanthoma elasticum Arterioscler Thromb Vasc Biol ( 2003 ) : 23: 836-841
[62] Pavlovic AM, Zidverc-Trajkovic J, Milovic MM Cerebral small vessel disease in pseudoxanthoma elasticum : three cases Can J Neurol Sci ( 2005 ) : 32: 115-118
[63] Le Corre Y, Naouri M, Martin L Pseudoxanthome élastique de l'enfant Ann Dermatol Venereol ( 2009 ) : 136: 552-555 quiz 551, 556
[64] Orssaud C, Roche O, Dufier J, Germain DP Visual impairment in pseudoxanthoma elasticum : a survey of 40 patients Ophthalmic Genet ( 2015 ) : 36: 327-332
[65] Kamoun B, Khlif H, Mseddi M Atteinte oculaire au cours du pseudoxanthome élastique Presse Med ( 2006 ) : 35: 779-783
[66] Oztas Z, Karadeniz C, Afrashi F Minor trauma resulting in subretinal haemorrhage with choroidal rupture : a case of subtle pseudoxanthoma elasticum in a child Clinical & Experimental Optometry ( 2016 ) : 99: 84-86
[67] Haroche J, Abla O Uncommon histiocytic disorders : Rosai-Dorfman, juvenile xanthogranuloma, and Erdheim-Chester disease Hematology Am Soc Hematol EducProgram ( 2015 ) : 2015: 571-578
[68] Niu L, Zhang C, Meng F Ocular juvenile xanthogranuloma Optom Vis Sci ( 2015 ) : 92: e126-e133
[69] Catt CJ, Hamilton GM, Fish J Ocular manifestations of Stevens-Johnson syndrome and toxic epidermal necrolysis in children Am J Ophthalmol ( 2016 ) : 166: 68-75
[70] Morales ME, Purdue GF, Verity SM Ophthalmic manifestations of Stevens-Johnson syndrome and toxic epidermal necrolysis and relation to SCORTEN Am J Ophthalmol ( 2010 ) : 150: 505-510 e1
[71] Sotozono C, Ueta M, Nakatani E Predictive factors associated with acute ocular involvement in Stevens-Johnson syndrome and toxic epidermal necrolysis Am J Ophthalmol ( 2015 ) : 160: 228-237 e2
[72] Sotozono C, Ueta M, Koizumi N Diagnosis and treatment of Stevens-Johnson syndrome and toxic epidermal necrolysis with ocular complications Ophthalmology ( 2009 ) : 116: 685-690
[73] Sharma N, Thenarasun SA, Kaur M Adjuvant role of amniotic membrane transplantation in acute ocular Stevens-Johnson syndrome : a randomized control trial Ophthalmology ( 2016 ) : 123: 484-491
M. Robert
On désigne par « maladies osseuses constitutionnelles » (MOC) un vaste groupe hétérogène d'environ 500 maladies classées en 40 groupes et caractérisées par une anomalie du cartilage et/ou de l'os. Le point d'entrée de certaines de ces maladies peut être une anomalie de l'œil (myopie congénitale), du nerf optique (neuropathie optique compressive) ou de l'orbite (déformation orbitaire). En outre, nombre d'entre elles requièrent après le diagnostic une surveillance ophtalmologique spécifique. On distingue au sein des MOC les ostéochondrodysplasies, les dysostoses et les anomalies de la structure osseuse. Les caractéristiques de l'atteinte oculaire des principales MOC affectant les yeux sont résumées dans le tableau 26-5. Nous détaillons ici les caractéristiques de l'ostéopétrose maligne et du syndrome d'Albers-Schönberg, les autres MOC étant traitées dans d'autres chapitres.
Tableau 26-5 – Caractéristiques de l’atteinte oculaire des principales maladies osseuses constitutionnelles affectant les yeux.
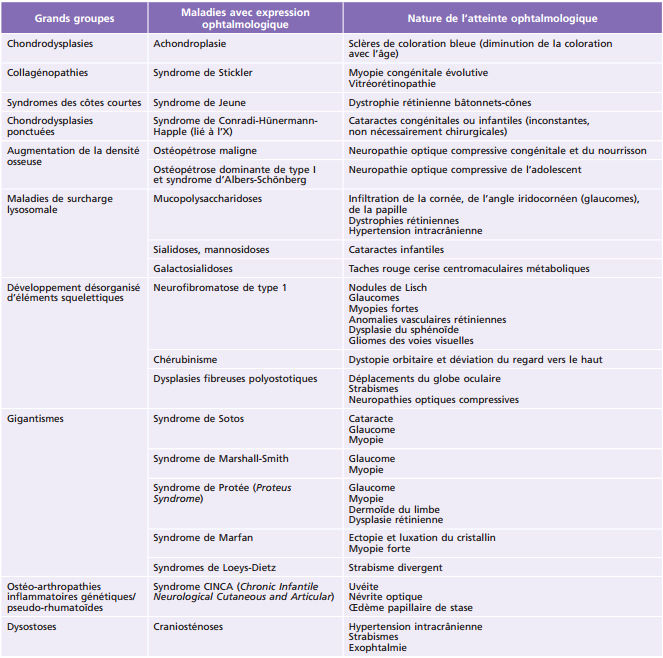
L'ostéopétrose maligne est une maladie autosomique récessive de la résorption osseuse dont le pronostic a été bouleversé par les progrès de l'immunologie pédiatrique : la greffe de moelle osseuse permet la survie de ces enfants et même, si elle est suffisamment précoce, la préservation d'une fonction visuelle. La maladie est hétérogène sur le plan génétique : mutations de T-cell immune regulator 1 (TCIRG1) dans environ 50 % des cas cependant. L'épaississement de la corticale des os aboutit à : d'une part, une déficience de la moelle osseuse, donc une pancytopénie et une hépatosplénomégalie par hématopoïèse extramédullaire; d'autre part, une dysmorphie faciale avec dilatation des veines du scalp et une compression des structures comprises dans des canaux osseux [1]. Il s'agit d'une des rares causes d'atrophie optique vraie congénitale ou du nourrisson.
L'atrophie optique peut survenir sur une papille hypoplasique du fait d'une atteinte in utero de la maladie; elle peut être accompagnée initialement d'un œdème papillaire de stase. Compte tenu des traitements actuels de la maladie, la décompression chirurgicale des nerfs optiques n'est en pratique guère réalisable dans un délai utile chez ces nourrissons. C'est le traitement de la maladie qui permet de préserver la fonction visuelle. Il est important de réaliser des électrorétinogrammes globaux chez ces enfants qui peuvent présenter, en association à l'atrophie optique, des dystrophies rétiniennes dont il est important de ralentir l'évolution par le port de verres teintés spécifiques [2].
Leur présentation est variable : scoliose, fractures, arthrose, ostéomyélite, compression des nerfs optiques dans les canaux optiques à l'origine d'une neuropathie optique (atrophie optique progressive parfois initialement accompagnée d'un œdème papillaire). On distingue deux phénotypes d'ostéopétrose dominante [3]. Le type I est caractérisé par une atteinte principalement crânienne; il n'existe pas d'augmentation du risque de fracture et la neuropathie optique est souvent le mode de révélation. La maladie résulte d'une activation de low-density lipoprotein receptor-related protein 5 (LRP5) et n'est plus considérée stricto sensu comme une ostéopétrose par certains auteurs [4]. L'examen clé est le scanner cérébral (fig. 26-22). Dans le type II, ou syndrome d'Albers-Schönberg, l'atteinte est, au contraire, plus diffuse et la neuropathie optique plus rare. La maladie résulte d'une mutation dans chloride voltagegated channel 7 (CLCN7). En cas de neuropathie optique, l'indication de chirurgie de décompression osseuse doit être posée au bon moment pour permettre une préservation de la fonction visuelle. Les enfants doivent donc être suivis à l'aide de la réalisation régulière d'un champ visuel. L'OCT et l'électrophysiologie visuelle sont souvent utiles dans le cadre de ce suivi, en faisant bien attention cependant aux effets opposés de l'œdème papillaire et de l'atrophie optique sur l'épaisseur de la couche des fibres ganglionnaires.
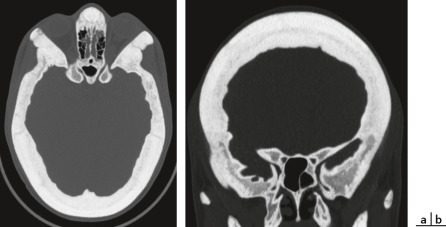
Fig. 26-22 Scanner cérébral non injecté dans une ostéopétrose dominante de type I montrant l’épaississement et la densification des os du crâne, l’étroitesse des canaux optiques et la dilatation des gaines des nerfs optiques.
a. Coupe axiale dans le plan neuro-oculaire. b. Coupe coronale passant par les canaux optiques.
[1] Sobacchi C, Schulz A, Coxon FP Osteopetrosis : genetics, treatment and new insights into osteoclast function Nat Rev Endocrinol ( 2013 ) : 9: 522-536
[2] Thompson DA, Kriss A, Taylor D Early VEP and ERG evidence of visual dysfunction in autosomal recessive osteopetrosis Neuropediatrics ( 1998 ) : 29: 137-144
[3] Andersen PE, Bollerslev J Heterogeneity of autosomal dominant osteopetrosis Radiology ( 1987 ) : 164: 223-225
[4] Bollerslev J, Henriksen K, Nielsen MF Autosomal dominant osteopetrosis revisited : lessons from recent studies Eur J Endocrinol ( 2013 ) : 169: R39-R57
G. Pech-Gourg, F. Audren, A. Sauer, E. Bui Quoc, M. Robert, D. Denis
G. Pech-Gourg
Ces tumeurs sont également traitées au chapitre 22.
Le craniopharyngiome est une tumeur épithéliale rare de bas grade histologique, se développant dans la région sellaire et suprasellaire à partir de résidus embryonnaires. Leur classement en tumeur de grade I dans la classification de l'Organisation mondiale de la santé (OMS) ne doit pas faire méconnaître la gravité potentielle de ces lésions, notamment en termes de retentissement cognitif, ophtalmologique et endocrinien.
L'exérèse totale de ces tumeurs a longtemps été considérée comme le seul garant d'une guérison définitive, mais malgré l'apport des techniques microchirurgicales, elle est loin d'être toujours sans risque et implique trop souvent des séquelles endocriniennes (panhypopituitarisme et diabète insipide) et neurocognitives [1]. Les nouvelles attitudes thérapeutiques combinées sont plus conservatrices, dans un souci de préservation fonctionnelle. Les taux de survie à 10 ans sont maintenant supérieurs à 90 % .
Les craniopharyngiomes représentent entre 1,2 et 4 % des tumeurs intracrâniennes chez l'enfant [2, 3]. Ils touchent surtout les enfants entre 7 et 13 ans avec une prédominance chez les garçons. Ils sont exceptionnellement découverts avant l'âge de 2 ans.
Sur le plan macroscopique le craniopharyngiome peut revêtir différentes formes : charnue, kystique ou mixte. Sur le plan microscopique, il s'agit d'une tumeur non gliale dérivant de tissus embryologiques épithéliaux dont l'origine reste controversée.
Les craniopharyngiomes sont, du fait de leur localisation, susceptibles de perturber les fonctions endocriniennes et visuelles. Leur volume au moment du diagnostic, mais aussi leurs rapports avec les structures anatomiques de la région sellaire et suprasellaire vont conditionner la présentation clinique.
Les différentes classifications topographiques tendent à décrire le développement de ces tumeurs par rapport à des limites anatomiques (fig. 26-23). Ainsi, les craniopharyngiomes peuvent être purement intrasellaires, suprasellaires ou plus rarement infrasellaires (ce qui sous-entend une effraction dans le sinus sphénoïde). Parmi les formes suprasellaires, on décrit le développement de la tumeur en fonction de sa position par rapport au chiasma optique : préchiasmatique ou rétrochiasmatique. Dans les formes les plus volumineuses et envahissantes, une extension supérieure peut soulever et déformer l'hypothalamus, voire l'envahir. Un développement vers le III ventricule peut avoir pour conséquence d'obstruer les foramens de Monro entraînant ainsi une hydrocéphalie avec HTIC.
En conséquence, le tableau clinique est fait de l'association, à des degrés variables, de troubles endocriniens d'origine hypothalamique et/ou hypophysaire, de troubles ophtalmologiques, de signes d'HTIC et de signes neurologiques focaux annexes.
Les signes révélateurs sont cependant plus volontiers une amblyopie et des signes d'HTIC chez l'enfant, un retard pubertaire ou un retard de croissance chez l'adolescent.
Les craniopharyngiomes ont une croissance relativement lente, c'est pourquoi les tableaux cliniques sont également d'installation progressive et insidieuse avec fréquemment une errance prédiagnostique.
Les signes visuels sont considérés comme signes d'appel dans 30 à 35 % des cas. Mais l'examen ophtalmologique systématique préopératoire est considéré comme anormal chez une majorité d'enfants, jusqu'à 96 % dans certaines séries pédiatriques [4]. L'amblyopie est souvent très sévère dès le premier examen puisque chez un enfant sur cinq, l'acuité visuelle est déjà nulle d'un côté.
Un tableau d'HTIC, très fréquemment révélateur chez l'enfant (60 à 75 % des cas), qu'il soit lié à une hydrocéphalie obstructive ou au volume de la lésion elle-même, donne des signes relativement aspécifiques de souffrance visuelle (baisse d'acuité visuelle bilatérale, œdème papillaire au fond d'œil, atrophie optique, diplopie horizontale par paralysie du VI).
Les anomalies du champ visuel sont conditionnées par les rapports de la tumeur avec les voies optiques et par le niveau de compression exercé (fig. 26-24).
Le craniopharyngiome suprasellaire se développe librement dans les espaces sous-arachnoïdiens de la base, à partir de son point de départ sur la tige pituitaire ou l'infundibulum. De son origine sur la ligne médiane, il peut se développer dans toutes les directions, à la fois dans le sens antéropostérieur et dans les axes latéraux :
- –s'il s'insinue entre les nerfs optiques et croît dans la région sous-frontale postérieure, il donnera une forme préchiasmatique se traduisant par une atteinte uni- ou bilatérale des nerfs optiques (fig. 26-24a);
- –s'il soulève le chiasma et étire les nerfs optiques, il donnera une forme sous-chiasmatique, avec classiquement une hémianopsie bitemporale (fig. 26-24b);
- –s'il se développe en arrière du bord postérieur du chiasma en écartant et comprimant les bandelettes optiques, il donnera une forme rétrochiasmatique qui, si elle est latéralisée, pourra donner un tableau d'hémianopsie latérale homonyme.
Le signe révélateur est le plus souvent une baisse uni- ou bilatérale de l'acuité visuelle, alors que l'altération du champ visuel se fait de façon progressive et est rarement perçue et difficilement exprimée par l'enfant.
De plus, le craniopharyngiome exerce à la fois une compression directe et des lésions indirectes par étirement sur les voies optiques. En conséquence, l'atteinte du champ visuel est rarement aussi stéréotypée que dans la théorie illustrée par les schémas. On retrouve généralement des amputations campimétriques asymétriques et anarchiques prédominant dans les hémichamps temporaux, beaucoup moins systématisées que dans les adénomes hypophysaires ou les méningiomes suprasellaires. Le champ visuel central est plus souvent atteint que le champ périphérique.
Les déficits du champ visuel sont plus difficiles à mettre en évidence chez l'enfant que chez l'adulte car l'étude campimétrique n'est réalisable qu'à partir de 6 ans à l'appareil de Goldmann, et après 8 à 9 ans pour les campimètres automatiques.
Fig. 26-23 Représentation schématique des éléments vasculonerveux de la région suprasellaire, pouvant être concernés par le développement d’un craniopharyngiome.
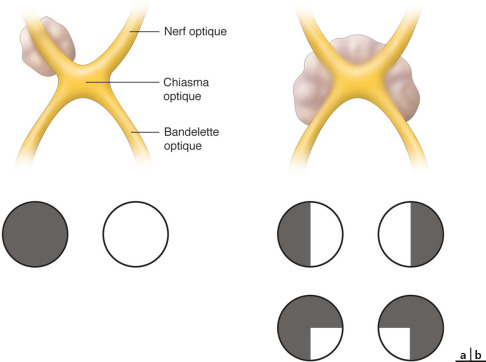
Fig. 26-24 Schématisation des anomalies campimétriques en fonction de la localisation tumorale.
Les altérations du champ visuel sont habituellement asymétriques et anarchiques, beaucoup moins systématisées que dans les adénomes hypophysaires ou les méningiomes suprasellaires. Néanmoins, un craniopharyngiome s’étendant en région pré- et sous-chiasmatique (région sellaire) se manifeste par une hémianopsie bitemporale ou des déficits temporaux par compression des fibres croisées ou nasales ; un volumineux craniopharyngiome rétrochiasmatique peut se manifester par un scotome central bilatéral par compression des fibres maculaires au niveau de leur croisement intrachiasmatique et par une hémianopsie latérale homonyme par compression d’une bandelette optique.
Le diagnostic est généralement effectué sans difficulté par le bilan d'imagerie, et confirmé par l'étude anatomopathologique.
Cette IRM peut être prescrite en première intention devant un déficit endocrinien ou une atteinte ophtalmologique. Elle décrit la tumeur charnue prenant le contraste de façon parfois hétérogène et le ou les kystes en hypersignal T2 (fig. 26-25). L'IRM permet de visualiser le chiasma (en utilisant le complexe artériel communicant antérieur comme repère anatomique) et ses déplacements ainsi que le troisième ventricule. L'IRM permet de définir le type de développement de la lésion et de choisir la voie d'abord chirurgicale.
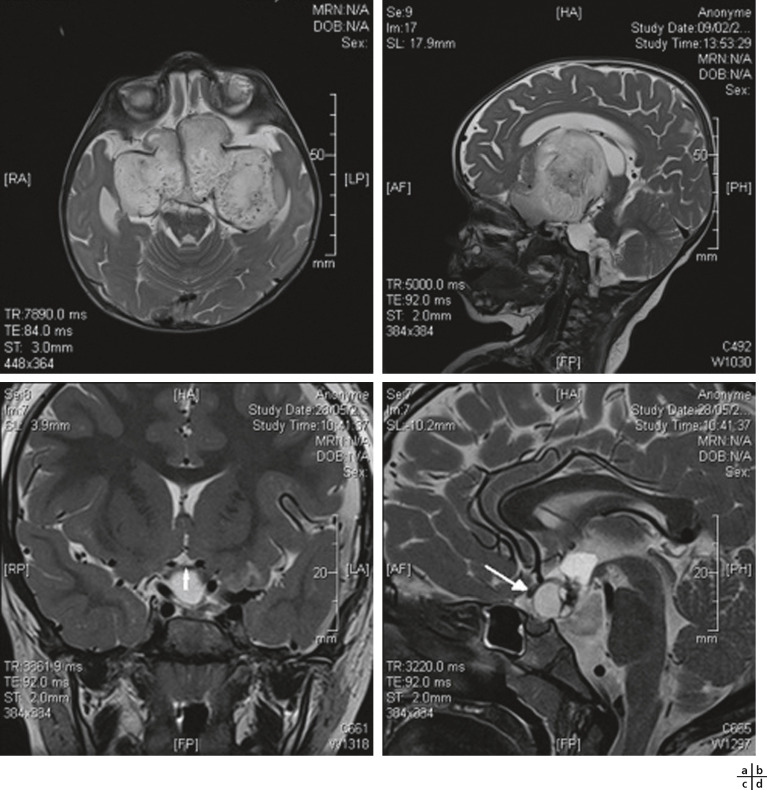
Fig. 26-25 Exemples d’IRM cérébrales d’enfants porteurs de craniopharyngiome.
a, b. IRM en séquence T2 axiale (a) et sagittale (b) d’un enfant âgé de 4 ans présentant un volumineux craniopharyngiome à expansion suprasellaire, révélé par un syndrome d’HTIC et présentant une baisse d’acuité visuelle bilatérale sévère. c, d. IRM en séquence T2 coronale (c) et sagittale (d) d’un enfant âgé de 6 ans présentant un craniopharyngiome suprasellaire à expansion postérieure. La position du chiasma optique (flèches) est déterminée à l’aide du repère anatomique que constitue l’artère communicante antérieure.
Examen de première intention devant une symptomatologie neurologique brutale, la tomodensitométrie (TDM) permet de faire le diagnostic d'une tumeur de la région sellaire et suprasellaire et d'une hydrocéphalie éventuelle. Elle garde son intérêt dans le diagnostic en complément de l'IRM en permettant de visualiser des calcifications intratumorales.
Les autres tumeurs suprasellaires de l'enfant constituent les diagnostics différentiels du craniopharyngiome. Les gliomes hypothalamo-chiasmatiques ont un retentissement visuel encore moins systématisé et sur l'IRM le chiasma est tumoral et non refoulé. Un germinome de la tige pituitaire sera évoqué devant un diabète insipide au diagnostic. Une atteinte bifocale pinéale et suprasellaire est possible et évocatrice. Enfin, certains macro-adénomes hypophysaires non sécrétants à extension suprasellaire peuvent représenter une difficulté diagnostique.
La prise en charge thérapeutique se doit d'être pluridisciplinaire. Il peut être nécessaire de traiter une hydrocéphalie en urgence devant un tableau d'HTIC. Mais un tableau de panhypopituitarisme pouvant entraîner des troubles de la conscience ne doit pas être méconnu et nécessite une substitution également en urgence (notamment minéralo-corticoïde).
La stratégie thérapeutique doit répondre à une triple nécessité : la confirmation histologique, la décompression des voies optiques et la préservation endocrinienne et neurocognitive. Les voies d'abord chirurgicales sont multiples (transsphénoïdale, ptérionale, sous-frontale, interhémisphérique, etc.) et doivent être choisies en fonction de la localisation de la tumeur mais aussi de l'objectif chirurgical préalablement décidé. Une exérèse complète ne constitue plus un impératif chirurgical grâce aux progrès des techniques de radiothérapie externe (conventionnelle, protonthérapie ou radiochirurgie stéréotaxique).
Alors que pendant plusieurs décennies, les neurochirurgiens tentaient de relever le défi d'une exérèse totale des craniopharyngiomes [5], l'accent est aujourd'hui mis sur la préservation de la qualité de vie des patients [6, 7]. En préservant l'hypothalamus, on préserve les fonctions neuropsychologiques et on diminue le risque de trouble de la satiété et donc d'obésité morbide postopératoire [8]. Il est souvent préférable de privilégier une attitude combinée conservatrice avec dans un premier temps une exérèse chirurgicale des parties kystiques et des portions charnues compressives en essayant de respecter les structures hypothalamiques et la tige pituitaire, puis dans un deuxième temps une radiothérapie ciblée sur le résidu charnu éventuel. Des réinterventions s'avèrent parfois nécessaires en cas de reprise évolutive des formes kystiques (notamment en cours de radiothérapie).
Une substitution hormonale sur les lignées hypophysaires déficitaires et sur un diabète insipide devra être effectuée en postopératoire et nécessitera un suivi endocrinien à vie.
Cette attitude conservatrice implique une surveillance régulière et prolongée des résidus tumoraux. Si l'IRM cérébro-hypophysaire est l'examen de référence de cette surveillance, le bilan ophtalmologique (acuité visuelle et champ visuel) occupe une place également prépondérante. En effet, une dégradation campimétrique peut être un signe plus sensible de reprise évolutive que l'IRM dans certains cas.
Ces tumeurs sont également traitées au chapitre 22.
Les gliomes des voies optiques sont des tumeurs rares (3 à 5 % des tumeurs cérébrales de l'enfant). L'association avec la N F1 est classique et l'incidence de la N F1 chez les patients porteurs d'un gliome des voies optiques varie entre 30 et 58 % . Ces tumeurs sont habituellement des astrocytomes pilocytiques et la régression de ces tumeurs a été décrite en particulier chez les enfants porteurs d'une NF1. L'exophtalmie, la baisse de l'acuité visuelle ou la cécité sont le mode de présentation usuel de ces tumeurs à évolution lente. L'imagerie par IRM permet l'étude des voies optiques. La chirurgie ne doit être proposée que pour les tumeurs évolutives et/ou avec une nécessité de confirmation histologique du diagnostic, en l'absence de NF1 connue par exemple, ou afin de restaurer la circulation du liquide céphalorachidien (LCR) dans les formes hypothalamo-chiasmatiques volumineuses. Les lésions du nerf optique intra-orbitaire sont opérées lorsqu'elles entraînent une exophtalmie sévère, avec vision non fonctionnelle ou amaurose [9]. La chimiothérapie doit être proposée en traitement de première intention pour les tumeurs évolutives avec retentissement visuel.
F. Audren, E. Bui Quoc, A. Sauer, M. Robert, D. Denis
La maladie de Steinert, ou dystrophie myotonique de type 1, est une dystrophie musculaire. Comme toutes les myopathies, elle entraîne des signes cliniques, biologiques, électromyographiques et histologiques résultant de l'atteinte des muscles striés. Elle comporte en outre des signes ophtalmologiques (cataracte, ophtalmoplégie et ptosis progressifs, anomalies de l'électrogenèse rétinienne et hypotonie oculaire), des troubles endocriniens (stérilité par hypogonadisme, diabète de type 2), des troubles cardiaques, du squelette et du système nerveux central [10].
C'est la plus fréquente des myopathies héréditaires (prévalence 1/8000); elle débute à l'adolescence et atteint l'adulte dans sa forme habituelle mais peut survenir chez l'enfant. C'est une maladie génétique à transmission autosomique dominante caractérisée par une répétition de triplets de bases d'ADN (acide désoxyribonucléique) : CTG = C (cytosine), T (thymine), G (guanine); le gène de cette maladie (DMPK ou dystrophia myotonica protein kinase) est localisé en 19q13. La maladie s'exprime variablement au-delà de 37 triplets et systématiquement si la répétition dépasse 50 triplets. Sa sévérité est très variable, allant des formes asymptomatiques aux formes congénitales de pronostic très sévère.
Comme dans toutes les maladies musculaires, il existe un syndrome myogène et des signes associés pouvant survenir à tout âge et s'installant de façon progressive. Le syndrome myogène associe un déficit moteur d'intensité variable, en général bilatéral et symétrique, à prédominance distale, sans trouble sensitif, associé à une amyotrophie de topographie variable.
Il existe trois formes de myotonie de Steinert :
- –une forme congénitale grave, avec à la naissance hypotonie majeure, insuffisance respiratoire sévère, troubles de la succion et de la déglutition, et espérance de vie limitée; cette forme congénitale est uniquement observée en cas de transmission maternelle, et ceci indépendamment du degré de gravité de l'atteinte chez la mère. En période anténatale, la réduction des mouvements actifs fœtaux et un hydramnios seront fortement évocateurs. La myotonie indétectable à la naissance sera recherchée chez la mère. Le diagnostic est confirmé par l'étude génétique moléculaire. Le pronostic est très sévère : décès dans près d'un quart des cas et, en cas de survie, déficit intellectuel fréquent;
- –une forme modérée classique de l’adulte ou de l’adolescent, avec myotonie et faiblesse musculaire, arythmie cardiaque qui conditionne le pronostic vital; habituellement diagnostiquée entre 10 et 30 ans. Le diagnostic est généralement facile, reposant sur l'association d'une faiblesse musculaire progressive, un faciès typique (calvitie précoce, grandes oreilles décollées) et des atteintes associées cardiaques (troubles du rythme : un bloc atrioventiculaire du 2 ou du 3 degré [11], endocriniennes, cérébrales, respiratoires, digestives;
- –une forme minime dans laquelle le seul signe significatif peut être la cataracte, typiquement polychromatique.
Les signes ophtalmologiques sont les suivants :
- –ptosis et troubles de motilité oculaire;
- –cataracte bilatérale et précoce : cataracte « myotonique » ;
- –pression oculaire inférieure à la normale [12, 13]; cette hypotonie peut se compliquer de choriorétinopathie hypotonique;
- –dysfonction rétinienne stationnaire : l'électrorétinogramme montre une diminution de l'onde b de la réponse mixte et de la réponse des cônes, avec une augmentation du temps de culmination de l'onde b [14]. L'association possible à des dystrophies maculaire n'est pas claire.
Les bilans sont les suivants :
- –biologique : dosage élevé des enzymes musculaires (créatine kinase, LDH, aldolases), dosage de la glycémie à jeun et de l'hémoglobine glyquée;
- –fonctionnel : électromyogramme recherchant des signes d'atrophie myogène et l'existence d'activités spontanées au repos;
- –histologique : biopsie musculaire pour un diagnostic positif sÛr mais non obligatoire;
- –biologie moléculaire : recherche par triplet repeat primed (TP)-polymerase chain reaction (PCR) de l'expansion des triplets CTG> 37;
- –explorations cardiaques et respiratoires : échographie, ECG, radiographie pulmonaire, Holter, explorations fonctionnelles respiratoires.
Il n'y a pas actuellement de traitement curatif. Le traitement est symptomatique. La prise en charge consiste en divers moyens médicamenteux et/ou techniques visant à améliorer la qualité de vie du sujet : éviction du froid et exercices musculaires pour améliorer la myotonie, kinésithérapie.
Des précautions spécifiques en cas d'anesthésie sont indispensables lorsqu'une intervention chirurgicale doit être réalisée.
On désigne par dysplasie septo-optique (syndrome de De Morsier) l'association d'une hypoplasie du nerf optique et d'un syndrome de la ligne médiane [15], Il convient d'y penser devant l'association insuffisance hypophysaire congénitale et anomalies oculaires (nystagmus, amblyopie profonde, hypoplasie des nerfs optiques et absence de septum pellucidum).
L'hypoplasie du nerf optique traduit un nombre réduit de fibres (voir chapitres 21 et 22), Elle peut s'associer à des déficits de la ligne médiane – hypoplasie du septum pellucidum, hypoplasie du 3 ventricule, intéressant le chiasma optique – et éventuellement une agénésie ou hypoplasie du corps calleux (fig. 26-26).
L'hypoplasie du nerf optique peut aussi s'accompagner de désordres endocriniens liés à un hypopituitarisme, souvent dÛ à déficit en hormone de croissance [16].
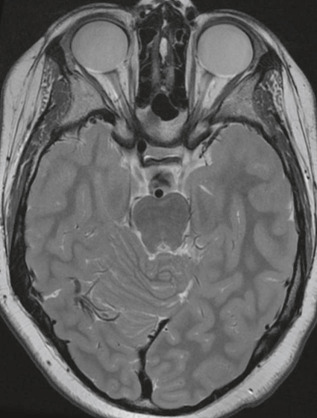
Fig. 26-26 Dysplasie septo-optique (IRM) : atrophie modérée des nerfs optiques prédominant à gauche ; agénésie du septum ; hétérotopie de la substance grise.
Rare chez l'enfant, la maladie de Basedow, maladie auto-immune, voit sa fréquence augmenter avec l'âge : exophtalmie, douleurs oculaires, larmoiement, strabisme par œdème des muscles oculomoteurs, tachycardie, hypertension artérielle (HTA), goître, Elle est confirmée par des taux sanguins élevés de thyroxine et nuls de thyroid stimulating hormone (TSH) et par la présence d'anticorps antithyroïdiens circulants dans le sang.
L'hypoparathyroïdie peut être post-chirurgicale ou idiopathique et peut donner une opacité cristallinienne sous-capsulaire postérieure bilatérale, parfois un œdème papillaire ou des troubles de la motricité oculaire. Ces troubles disparaissent après normalisation de la calcémie et de la phosphorémie [17].
Le syndrome de Laurence Moon-Biedl associe une surcharge pondérale, une insuffisance gonadique, un retard développemental et une rétinopathie pigmentaire.
Le syndrome de Wolfram associe diabète insipide, diabète sucré, atrophie optique et surdité.
Complication majeure du diabète insulino-dépendant, la rétinopathie, rare avant 3 ans d'évolution chez l'enfant, revêt les mêmes stades que chez l'adulte : rétinopathie non proliférante (œdème, exsudats) ou proliférante (néovaisseaux intrarétiniens, hémorragies intravitréennes, décollement de rétine).
On préconise une angiofluorographie après 3 ans d'évolution, puis un contrôle tous les 2 ans, sauf si des lésions apparaissent accélérant les contrôles. Il convient d'équilibrer au plus vite et au mieux la glycémie.
[1] Müller HL Childhood craniopharyngioma Pituitary: ( 2013 ) : 16: 56-67
[2] Bauchet L, Rigau V, Mathieu-Daudé H Société française de neurochirurgie pédiatrique; Société française de neurochirurgie; Société française de neuropathologie; Association des neuro-oncologues d'expression française. Clinical epidemiology for childhood primary central nervous system tumors J Neurooncol: ( 2009 ) : 92: 87-98
[3] Bunin GR, Surawicz TS, Witman PA The descriptive epidemiology of craniopharyngioma J Neurosurg: ( 1998 ) : 89: 547-551
[4] Defoort-Dhellemmes S, Moritz F, Bouacha I, Vinchon M Craniopharyngioma : ophtalmological aspects at diagnosis J Pediatr Endocrinol Metab: ( 2006 ) : 19: 321-324
[5] Yasargil MG, Curcic M, Kis M Total removal of craniopharyngiomas. Approaches and long-term results in 144 patients J Neurosurg: ( 1990 ) : 73: 3-11
[6] Sainte-Rose C, Puget S, Wray A Craniopharyngioma : the pendulum of surgical management Childs Nerv Syst: ( 2005 ) : 21: 691-695
[7] Ogawa Y, Kawaguchi T, Tominaga T Outcome and mid-term prognosis after maximum and radical removal of craniopharyngiomas with the priority to the extended transsphenoidal approach – A single center experience Clin Neurol Neurosurg: ( 2014 ) : 125C: 41-46
[8] Elowe-Gruau E, Beltrand J, Brauner R Childhood craniopharyngioma : hypothalamus-sparing surgery decreases the risk of obesity J Clin Endocrinol Metab: ( 2013 ) : 98: 2376-2382
[9] Lena G, Pech-Gourg G, Scavarda D Gliome du nerf optique chez l'enfant Neurochirurgie: ( 2010 ) : 56: 249-256
[10] Brook JD, McCurrach ME, Harley HG Molecular basis of myotonic dystrophy : expansion of a trinucleotide (CTG) repeat at the 3' end of a transcript encoding a protein kinase family member Cell: ( 1992 ) : 68: 799-808
[11] Groh WJ, Groh MR, Saha C Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1 N Eng J Med: ( 2008 ) : 358: 2688-2697
[12] Raby O, Bonsch M [Ocular hypotonia and retinal injuries in Steinert disease. Angiographic aspects] JFO: ( 1986 ) : 9: 543-552
[13] Garcia Filho CA, Prata TS, Sousa AK Intraocular pressure, corneal thickness, and corneal hysteresis in Steinert's myotonic dystrophy Arq Bras Oftalmol: ( 2011 ) : 74: 161-162
[14] Segal BS The retinopathy of dystrophia myotonia Steinert Metab Pediatr Sys Ophthalmol: ( 1986 ) : 9: 585-587
[15] De Morsier G Études sur les dysraphies cranio-encéphaliques III. Agénésie du septum pellucidum avec malformation du tractus optique. La dysplasie septo-optique Schweiz Arch Neurol Psychiatr: ( 1956 ) : 77: 267-272
[16] Fink C, Borchert M, Simon CZ, Saper C Hypothalamic dysfunction without hamartomas causing gelastic seizures in optic nerve hypoplasia J Child Neurol: ( 2015 ) : 30: 233-237
[17] Chauvin MC. Manifestations ophtalmologiques de l’hypoparathyroidie primitive de l’enfant. À propos de 16 observations [thèse de médecine]. Paris VI ; 1987.
C. Orssaud, C. Seghir, M. Robert
Les maladies métaboliques résultent de la présence d'un déficit enzymatique (fig. 26-27). Ce déficit résulte d'une mutation récessive (transmission récessive autosomique ou RA) dans l'immense majorité des cas, d'où la fréquence de ces maladies dans les populations consanguines. Les signes sont la conséquence d'une intoxication par accumulation de A (excès de A) et/ou d'une carence par défaut de B (manque de B). En fonction des cas, les signes peuvent être présents à la naissance ou au contraire apparaître à un âge variable. Toute perte ou dégradation d'une fonction acquise précédemment chez un nourrisson ou un enfant doit faire évoquer une maladie métabolique. La recherche de cette notion de régression est un élément fondamental de l'interrogatoire en ophtalmologie pédiatrique.
L'ophtalmopédiatre est un acteur essentiel de la prise en charge de ces maladies : il en fait souvent le diagnostic, à partir d'un symptôme visuel; il oriente le diagnostic étiologique en mettant en évidence un signe spécifique permettant de cibler les recherches; il apprécie l'efficacité du traitement en suivant l'évolution de l'atteinte oculaire.
Plutôt qu'une classification génétique ou biochimique, nous avons ici fait le choix de présenter ces maladies en fonction de leur principale expression ophtalmologique et sous forme de tableaux synthétiques dès lors que la classification s'y prêtait :
- –atteintes palpébrales;
- –atteintes de la cornée et du segment antérieur de l'œil;
- –atteintes du pôle postérieur avec : tache rouge cerise centromaculaire; dystrophie rétinienne; dystrophie maculaire cristalline; infiltration papillaire; hémorragie papillaire;
- –atteintes oculomotrices.
Cette classification demeure sans doute la plus lisible pour l'ophtalmologiste; elle est cependant en grande partie artificielle, du fait que l'expression de ces maladies est généralement très large : plusieurs organes sont atteints; un même organe – l'œil – pouvant être atteint dans l'ensemble de ses composantes sus-nommées. Ainsi, par exemple, avons-nous choisi de classer la maladie de Fabry et les mucopolysaccharidoses au sein des atteintes du segment antérieur en raison de leur atteinte la plus souvent évidente, alors que la rétine et le nerf optique sont aussi affectés par ces maladies.

Fig. 26-27 Principe schématique simplifié des erreurs innées du métabolisme.
La protéinose lipoïde d'Urbach-Wiethe se caractérise par une raucité de la voix précoce et d'aggravation progressive, une coloration jaunâtre de la peau, une blépharose moniliforme (accumulation de petits nodules jaunâtres en chapelet sur les bords libres des quatre paupières), une infiltration de la langue et des cordes vocales [1], Des troubles respiratoires et une épilepsie peuvent être présents, Le scanner cérébral montre généralement la présence de calcifications des amygdales et des lobes temporaux, L'ensemble résulte de dépôts de matériel hyalin dans différents tissus, Le gène impliqué est ECM1 (1q21); la transmission est récessive autosomique; la prévalence maximale est en Europe du Nord,
La maladie de Fabry (MF) est une glycosphingolipidose due à un déficit de l’alpha-galactosidase A (aGA), enzyme lysosomal homodimérique [2], codé par le gène GLA (Xq21.3). L’accumulation de globotriaosylcéramide (Gb3) et, à un moindre degré, de galabiosylcéramide dans les cellules vasculaires endothéliales de l’ensemble de l’organisme est responsable de complications ischémiques au niveau cérébral, rénal et cardiaque. Le décès des patients survient entre la quatrième et la sixième décennie [3]. La MF est transmise selon un mode lié au chromosome X. Les hommes, homozygotes pour le gène muté, n’ont aucune activité de l’aGA et développent une forme sévère de la maladie [2]. Les femmes, hétérozygotes, gardent une activité enzymatique. Du fait du phénomène de lyonisation du chromosome X, elles développent des symptômes plus ou moins sévères de la MF [3].
Les manifestations générales apparaissent dans l’enfance et associent angiokératomes et acroparesthésies. Des complications vasculaires, insuffisance rénale et cardiaque et accidents neurologiques, dues à l’accumulation de Gb3 dans les parois vasculaires apparaissent à l’âge adulte.
De nombreuses manifestations ophtalmologiques sans retentissement visuel ont été rapportées lors de la MF [4]. La cornée verticillée (voir fig. 26-2) est retrouvée chez la plupart des hommes atteints ainsi que chez les femmes hétérozygotes, c’est pourquoi elle constitue un marqueur génétique de cette affection [5]. Cette cornée verticillée apparaît précocement au cours de la vie et des anomalies histologiques cornéennes ont été retrouvées chez un foetus porteur de la mutation [6]. Avec l’âge, elle évolue vers un haze diffus chez les patients homozygotes [4]. Les inclusions siègent au niveau de la couche des cellules basales de l’épithélium cornéen et au niveau du stroma antérieur [7]. Les télangiectasies conjonctivales peuvent également être retrouvées chez les grands enfants. La tortuosité des vaisseaux rétiniens, fréquente chez les patients atteints de MF, n’est généralement retrouvée que chez l’adulte. Il en est de même des opacités cristalliniennes triangulaires équatoriales ou postérieures [4].
Il existe un traitement par enzymologie recombinante qui a démontré sa relative efficacité et sa sécurité [8].
L’atteinte cornéenne peut être l’expression principale du syndrome de Richner-Hanhart, ou tyrosinémie de type 2, ou tyrosinémie oculocutanée. C’est une maladie rare de transmisssion récessive autosomique secondaire à une mutation dans le gène de la tyrosine aminotransférase (TAT) en 16q22.1. L’atteinte cutanée consiste en une kératodermie palmoplantaire douloureuse. L’atteinte oculaire résulte d’une atteinte de l’épithélium cornéen superficiel : il existe typiquement une kératopathie en chaînette, caractéristique, avec élevures moniliformes de l’épithélium ne prenant pas la fluorescéine entre les phases aiguës douloureuses photophobes (fig. 26-28), se rompant régulièrement pour prendre un aspect parfois pseudo-dendritique généralement confondu avec une kératopathie épithéliale herpétique. Une atteinte neurologique est souvent associée avec retard mental modéré. Le traitement consiste en un régime alimentaire pauvre en phénylalanine et en tyrosine [9].
La cystinose résulte d’un défaut de transport de la cystine hors des lysosomes, à l’origine d’une accumulation de cystine dans différents organes. Il en existe trois formes : les deux premières sont des maladies systémiques ; l’atteinte ophtalmologique permet une confirmation immédiate du diagnostic suspecté, est invalidante et requiert un traitement spécifique par collyre au chlorhydrate de cystéamine en sus du traitement systémique ; la forme adulte se résume à la kératopathie cristalline (voir fig. 26-1 et tableau 26-6) [10, 11].
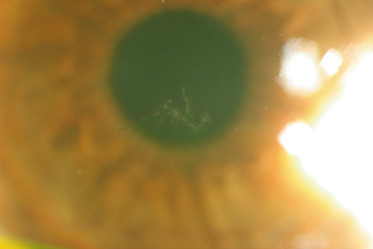
Fig. 26-28 Kératopathie en chaînette dans un syndrome de Richner-Hanhart.
La maladie de Wilson est abordée avec les atteintes de l’oculomotricité en fin de chapitre. L’anneau de Kayser-Fleischer ne se voit jamais « par hasard » : il convient de rechercher le liseré cuivré caractéristique à la lampe à fente, au niveau de la membrane de Descemet, à la jonction sclérocornéenne et d’abord à midi dans les formes débutantes (fig. 26-29).
La surcharge des maladies ainsi désignées peut atteindre la cornée. Le plus souvent, la protéine accumulée est opaque, en sorte que la cornée perd progressivement sa transparence et s’épaissit (fig. 26-30). Cette kératopathie stromale peu spécifique doit faire évoquer une maladie de surcharge : l’ophtalmologiste s’attachera à rechercher d’autres atteintes ophtalmologiques et générales pouvant orienter le diagnostic (tableau 26-7).
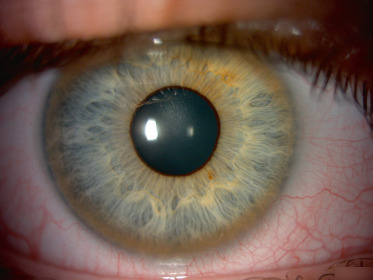
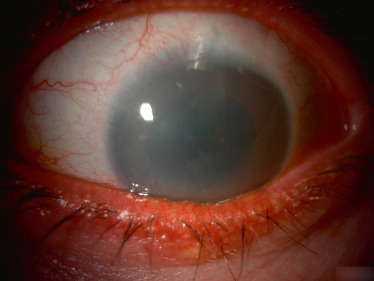
Fig. 26-30 Opacification stromale diffuse dans une mucopolysaccharidose.
Tableau 26-7 Maladies métaboliques pouvant s'associer à une infiltration stromale diffuse.
AR : autosomique récessive ; LCAT : lécithine cholestérol acyl transférase ; MPS : mucopolysaccharidoses.
Les mucopolysaccharidoses désignent un vaste ensemble de maladies ayant en commun l’accumulation de glycosaminoglycanes, anciennement appelés mucopolysaccharides. Celle-ci résulte d’un déficit en enzyme lysosomale et aboutit à une surcharge progressive de la plupart des tissus et organes. Les manifestations ophtalmologiques conduisent souvent au diagnostic et sont importantes à connaître, ainsi que les manifestations systémiques associées, notamment la dysmorphie faciale caractéristique « en gargouille » , car leur présence permettra un diagnostic précoce, d’autant plus crucial que les possibilités thérapeutiques sont désormais passées du domaine de la recherche à celui de la pratique clinique – greffes de cellules souches hématopoïétiques et de plus en plus traitements enzymatiques substitutifs qui permettent de ralentir ou stopper l’évolution de la maladie tableaux 26-8 et 26-9) [12].
Tableau 26-8 Principales manifestations cliniques (hors atteinte ophtalmologique) des mucopolysaccharidoses.
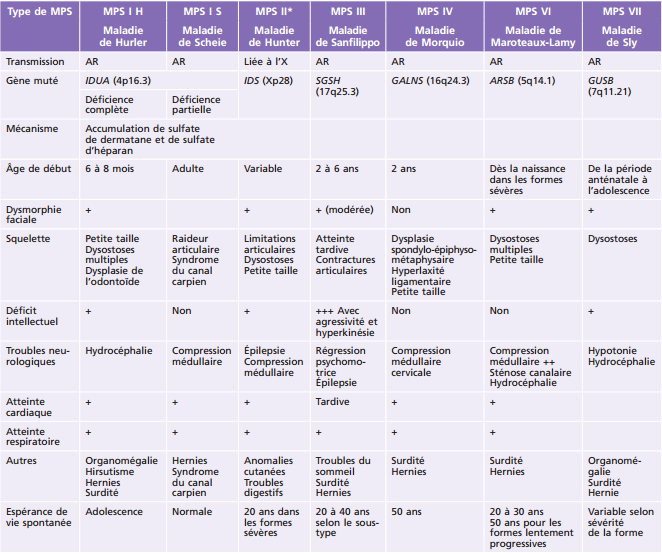
* Description de la forme sévère ; dans les formes modérées, l’intelligence est conservée, les dysostoses et la dysmorphie faciale sont moins marquées, et l’espérance de vie peut atteindre 60 ans. AR : autosomique récessive ; MPS : mucopolysaccharidoses.
Tableau 26-9 Degré de sévérité des différentes atteintes ophtalmologiques selon le type de MPS d'après Ashworth et al., 2006 [12]
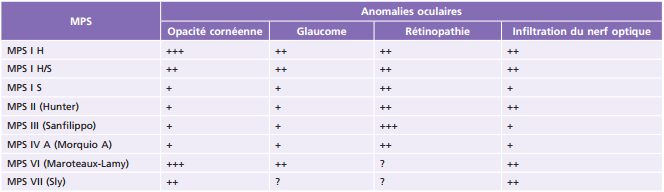
+ : léger, ++ : modéré, +++ : sévère, ? : non décrit ; MPS : mucopolysaccharidoses.
L’alcaptonurie, ou ochronose, est le modèle historique des maladies métaboliques. Elle résulte d’une accumulation d’acide homogentisique et de son produit d’oxydation, l’acide benzoquinone acétique. Le gène muté, HGD, est situé en 3q13. Le diagnostic est parfois porté à la naissance sur l’aspect foncé des urines après leur émission. Dans le cas contraire, le diagnostic est souvent tardif, à l’âge adulte, posé sur les signes rhumatologiques ou, à l’extrême, orthopédiques de l’arthropathie ochronotique, devant la constatation peropératoire d’une cavité articulaire noire à l’occasion d’une pose de prothèse articulaire. Il existe également une pigmentation de l’hélix et de la peau en regard des articulations. Sur le plan ophtalmologique, les signes sont malheureusement méconnus ; ils permettraient souvent, à l’occasion d’un examen de routine, un diagnostic plus précoce de cette maladie pour laquelle un traitement est à l’étude. Ils consistent en l’apparition, à l’adolescence ou chez l’adulte, de taches limbiques en gouttelettes brunâtres, spécifiques de la maladie, parfois d’une pigmentation ponctuée de la conjonctive en regard, suivie par l’apparition d’une coloration gris bleuté de la sclérotique en regard de l’insertion des muscles droits horizontaux (fig. 26-31). Il convient de s’assurer de l’absence de glaucome ou d’astigmatisme associés [13].
La découverte d’une homocystinurie se fait souvent via l’ophtalmologiste, devant l’association chez un enfant d’un astigmatisme et d’une myopie (de mécanisme mixte : d’indice, puis également axile) qui s’aggravent progressivement. Une dilatation pupillaire permet de mettre en évidence une ectopie cristallinienne : il est important de bien comparer l’épaisseur du cristallin dans chaque quadrant après dilatation, en fente fine, afin de mettre en évidence une ectopie débutante. Cette ectopie peut consister en un déplacement du cristallin dans n’importe quelle direction. Les autres signes de la maladie incluent : un morphotype marfanoïde, des anomalies squelettiques (genu valgum, pied creux, dolichosténomélie, pectus excavatum ou carinatum, cyphose ou scoliose, ostéoporose), un déficit intellectuel variable d’aggravation lentement progressive et inconstamment des troubles psychiatriques. La mortalité résulte des thromboses, artérielles et veineuses, favorisées par les anesthésies générales, d’où la règle de ne jamais opérer un patient d’ectopie du cristallin avant d’avoir éliminé une homocystinurie. La maladie, de transmission récessive autosomique, résulte d’une mutation dans le gène CBS (21q22.3). Il existe des traitements efficaces qui permettent d’éviter le développement des signes et des complications de la maladie, mais pas leur régression (notamment le retard mental), d’où l’importance d’un diagnostic le plus précoce possible [14, 15].
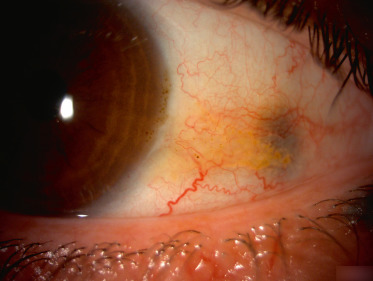
Fig. 26-31 Taches limbiques en gouttelettes brunâtres, pigmentation ponctuée de la conjonctive en regard et coloration gris bleuté de la sclérotique en regard de l'insertion du muscle droit latéral dans le cadre d'une alcaptonurie (ochronose).
La découverte d’une cataracte congénitale bilatérale ou d’une cataracte infantile entraîne une démarche étiologique spécifique, clinique, qui est exposée dans le chapitre 13. Parmi les causes possibles, l’on retrouve certaines maladies métaboliques d’autant plus importantes à diagnostiquer que d’une part, elles s’associent à des atteintes extra-oculaires et d’autre part, elles bénéficient parfois – et de plus en plus – de traitements permettant d’en stopper l’évolution. La nature de la cataracte est rarement informative. Le diagnostic peut souvent être suspecté dès l’inspection ou l’interrogatoire [16, 17]. Dans les formes du nourrisson limitées à une opacité sous-capsulaire postérieure (fig. 26-32), il est important de poser le diagnostic d’une anomalie du métabolisme du galactose avant d’opérer la cataracte, car le traitement médical de cette anomalie peut entraîner la régression, voire dans certains cas la disparition de la cataracte.
Dans les formes pédiatriques, le diagnostic de xanthomatose cérébro-tendineuse ne doit pas être retardé, car la cataracte peut être le premier signe d’une maladie désormais curable à condition d’être diagnostiquée précocement.
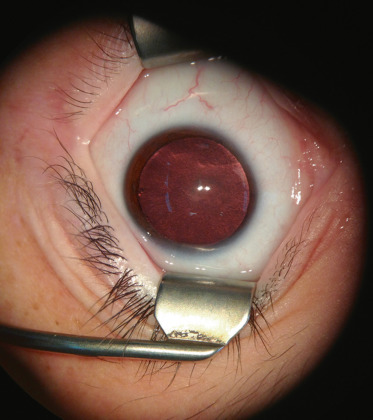
Fig. 26-32 Cataracte sous-capsulaire postérieure dans une galactosémie.
La question du bien-fondé d’un bilan systématique à réaliser devant toute cataracte congénitale ou pédiatrique est débattue. Hormis la réalisation systématique d’une échographie cardiaque préopératoire afin d’éliminer une cardiomyopathie hypertrophique dans le cadre d’un exceptionnel syndrome de Sengers ( fig. 26-33), un bilan systématique serait à la fois coûteux et peu informatif ; un examen et une surveillance cliniques guidés par la connaissance des diagnostics à rechercher sont par conséquent la règle aujourd’hui dans de nombreux centres spécialisés (tableaux 26-10 à 26-12).
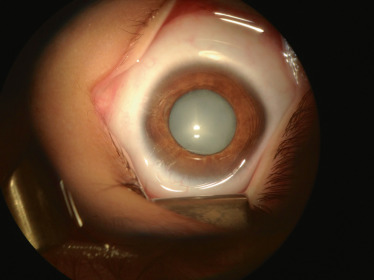
Fig. 26-33 Cataracte blanche dans un syndrome de Sengers.
Tableau 26-10 Maladies métaboliques pouvant s'associer à une cataracte.

Tableau 26-11 Désordres du métabolisme des glucides pouvant s'associer à une cataracte.
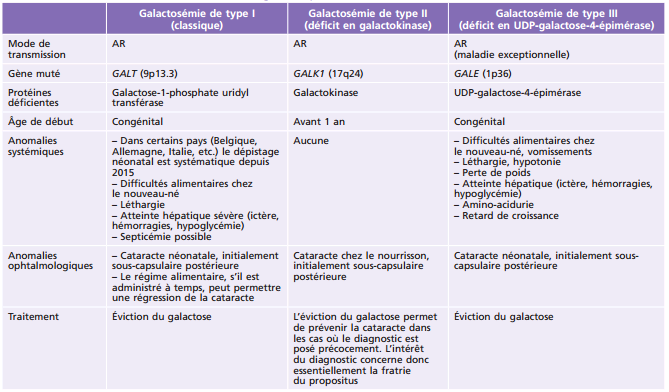
AR : autosomique récessive.
Tableau 26-12 Autres maladies métaboliques pouvant s'associer à une cataracte.
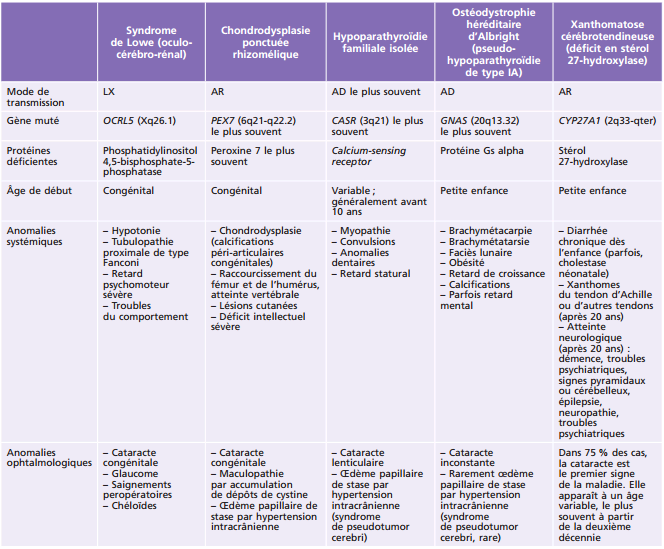
AD : autosomique dominante ; AR : autosomique récessive ; LX : liée à l’X.
Par dystrophie rétinienne, nous entendons tout processus pathologique principalement caractérisé par une dégénérescence des photorécepteurs : celle-ci peut concerner :
- –les bâtonnets puis les cônes et l'on parle alors de dystrophies bâtonnets-cônes (fig. 26-34), anciennement appelées « dégénérescences tapéto-rétiniennes » ou « rétinopathies pigmentaires, ou encore « rétinite pigmentaire » ; par définition, à l'électrorétinogramme (ERG) global, des réponses des bâtonnets, puis également celles des cônes, sont altérées;
- –ou à l'inverse, plus rarement, les cônes puis les bâtonnets et l'on parle alors de dystrophies cônes-bâtonnets (fig. 26-35); il existe alors d'abord cliniquement une maculopathie, mais l'ERG montre que le processus n'est pas limité aux cônes maculaires et concerne bien l'ensemble des cônes de la rétine avant d'affecter également les bâtonnets.
Devant toute dystrophie rétinienne de l'enfant, on doit se poser la question « syndromique, ou isolée » . Il n'est pas rare que la découverte d'une dystrophie rétinienne conduise à un diagnostic de maladie métabolique, potentiellement curable. Certaines présentations cliniques et électrophysiologiques sont assez caractéristiques et orientent le clinicien vers le diagnostic; il est donc important de les connaître. Ainsi, l'association d'une dystrophie rétinienne à une atrophie optique peut orienter vers une maladie du peroxysomale (fig. 26-36). Ainsi, les acidémies méthylmaloniques avec homocystinurie par déficit en cobalamine C, parmi de nombreuses autres présentations possibles, sont l'une des rares causes de dystrophie rétinienne cônes-bâtonnets avec une maculopathie présente cliniquement dès la naissance (fig. 26-37). Ainsi, une électronégativité des réponses de l'ERG global devant dystrophie rétinienne associée à des signes neurologiques débutants oriente vers une atteinte de la rétine interne, assez évocatrice de céroïde lipofuschinose neuronale juvénile. Dans d'autres cas, l'atteinte clinique rétinienne est peu spécifique, avec une atrophie rétinienne et des accumulations de pigment en mottes plutôt qu'en ostéoblastes chez l'enfant; il est donc également capital de rechercher les signes extra-ophtalmologiques de ces maladies. À l'inverse, devant une suspicion de maladie métabolique de l'enfant, l'ophtalmologue sera amené à rechercher une dystrophie rétinienne [14, 15]. Enfin, après le diagnostic de certaines maladies métaboliques, il faudra prévenir et surveiller l'apparition, puis prendre en charge une dystrophie rétinienne associée. Dans ces trois cas de figure, la place des investigations électrophysiologiques est souvent cruciale (tableaux 26-13 à 26-16).
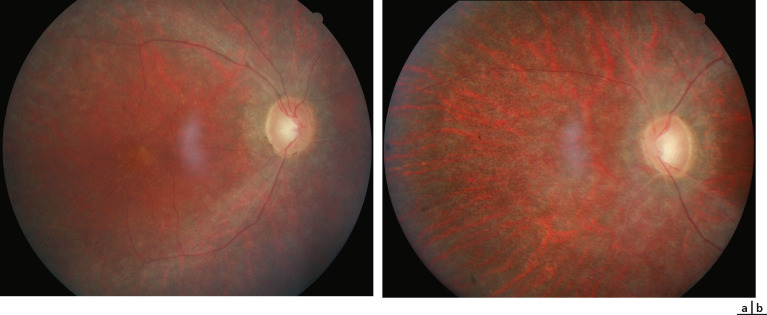
Fig. 26-34 Dystrophie rétinienne bâtonnets – cônes métabolique.
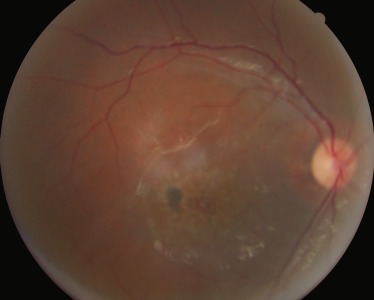
Fig. 26-35 Dystrophie rétinienne cônes – bâtonnets métabolique.
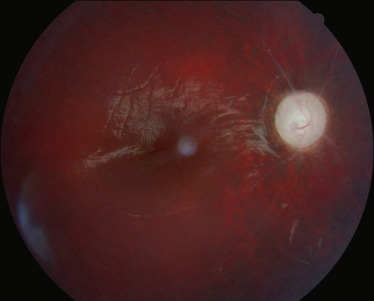
Fig. 26-36 Dystrophie rétinienne mixte et atrophie optique dans le cadre d'une maladie peroxysomale.
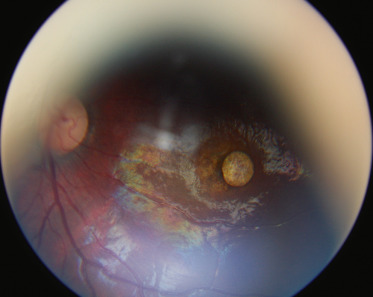
Fig. 26-37 Maculopathie néonatale dans une acidémie méthylmalonique avec homocystinurie par déficit en cobalamine C avec dystrophie rétinienne cônes-bâtonnets.
C'est volontairement que nous ne classons pas ces rétinopathies dans les dystrophies rétiniennes, car généralement les réponses de l'ERG global sont longtemps préservées; l'accumulation des cristaux concerne principalement l'épithélium pigmentaire. Bien sÛr, les rétinopathies cristallines résultent essentiellement de causes toxiques; rarement toutefois, elles peuvent se voir dans au moins deux maladies métaboliques : la cystinose (voir plus haut; l'atteinte cornéenne est alors toujours présente et la rétinopathie ne pose guère de question diagnostique) et le groupe des hyperoxaluries primitives.
Tableau 26-13 Principales maladies métaboliques pouvant s'associer à une dystrophie rétinienne.
CLN : céroïdes lipofuscinose neuronales.
Tableau 26-14 Principales manifestations des maladies peroxysomales.
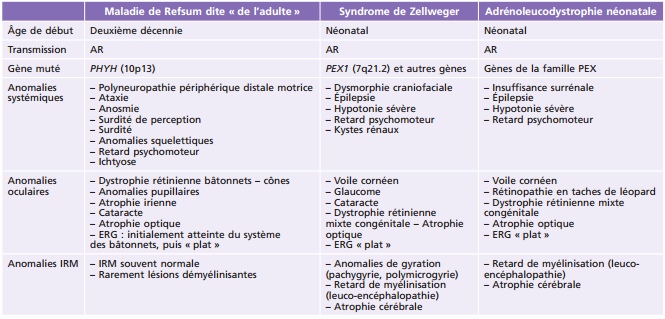
AR : autosomique récessive ; ERG : électrorétinogramme ; IRM : imagerie par résonance magnétique.
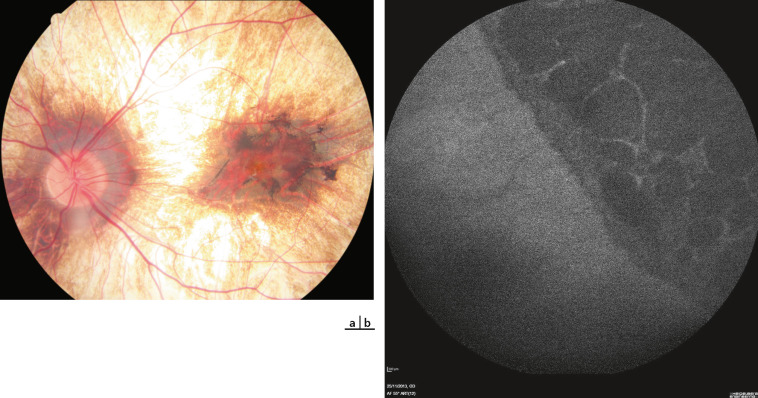
Fig. 26-38 Stades 3 avancé (a) et 4 (b) de la dystrophie choriorétinienne des déficits en 3-hydroxyacyl-CoA déshydrogénase des acides gras à chaîne longue (L-CHAD).
a. Rétinophotographie en couleurs. b. Cliché en autofluorescence, montrant la ligne de démarcation entre une zone de rétine normale en périphérie et une zone d’atrophie choriorétinienne.
Tableau 26-15 Principales manifestations des céroïdes lipofuscinoses neuronales.
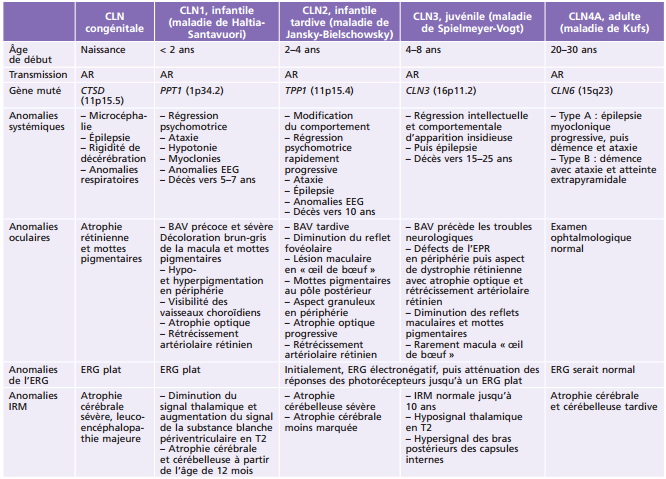
AR : autosomique récessive ; BAV : baisse de l’acuité visuelle ; CLN : céroïdes lipofuscinoses neuronales ; EEG : électro-encéphalographie ; EPR : épithélium pigmentaire de la rétine ; ERG : électrorétinogramme ; IRM : imagerie par résonance magnétique.
Tableau 26-16 Principales manifestations du déficit en L-CHAD et de l'acidémie méthylmalonique avec homocystinurie par déficit en cobalamine C.
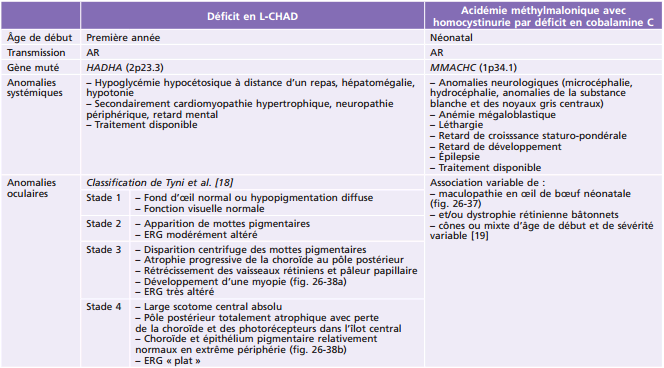
AR : autosomique récessive ; ERG : électrorétinogramme ; L-CHAD : 3-hydroxyacyl-CoA déshydrogénase des acides gras à chaîne longue.
Il existe trois types (1, 2 et 3) d'hyperoxalurie primitive. La rétinopathie signe l'atteinte systémique de la maladie et elle se voit dans environ un tiers des types 1, essentiellement les formes à début précoce. Elle se caractérise par un tableau assez spécifique avec successivement : accumulation diffuse de cristaux sous-rétiniens d'oxalose, boucles brunes sous-rétiniennes, fibrose sous-maculaire (fig. 26-39 et tableau 26-17) [20].
L'acidurie glutarique de type 1 est une pathologie qui doit être bien connue des neurochirurgiens et des ophtalmopédiatres : elle constitue en effet un diagnostic différentiel classique et essentiel du syndrome du bébé secoué, notamment dans les formes à début insidieux sans encéphalopathie métabolique aiguë évocatrice [21]. Une analyse attentive de la clinique et surtout de l'imagerie permet alors néanmoins de suspecter le diagnostic [22, 23]. L'accumulation d'acide glutarique, d'acide 3-hydroxyglutarique, d'acide glutaconique et de glutaryl-carnitine résulte en une dilatation des espaces sous-arachnoïdiens, qui distendent les veines ponts qui les traversent. Celles-ci peuvent se rompre, par exemple à l'occasion d'un traumatisme minime et entraîner un hématome sous-dural (tableau 26-18).
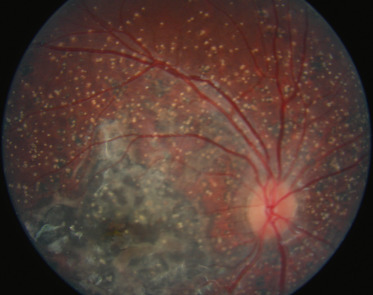
Fig. 26-39 Rétinopathie cristalline avec boucles brunes et fibrose sous-maculaire dans une hyperoxalurie primitive de type 1.
Tableau 26-17 Principales manifestations de l'hyperoxalurie primitive de type 1.
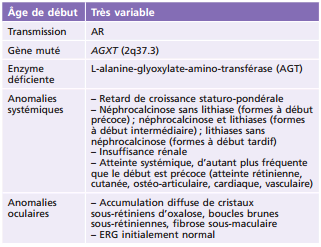
AR : autosomique récessive ; ERG : électrorétinogramme.
La découverte d'une tache rouge cerise centromaculaire métabolique (fig. 26-40) survient généralement au cours de l'examen au chevet d'un enfant présentant une atteinte neurologique sévère. Elle permet d'orienter les recherches vers certaines maladies de surcharge lysosomale. Cette tache rouge cerise résulte de l'accumulation de lipides dans la rétine, de ce fait épaissie sauf au niveau de la fovéa. Sa disparition secondaire à la dissipation du matériel est possible, laissant alors place à une atrophie optique séquellaire (tableaux 26-19 à 26-24) [24].
Tableau 26-18 Principales manifestations de l'acidurie glutarique de type 1 (déficit en glutaryl-coenzyme A déshydrogénase).
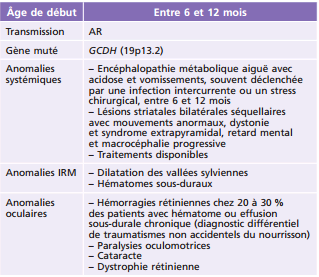
AR : autosomique récessive ; IRM : imagerie par résonance magnétique.
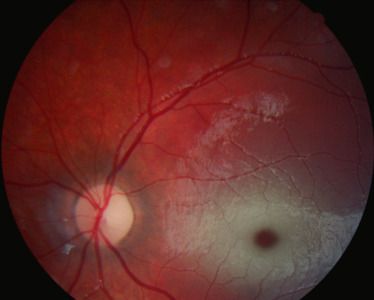
Fig. 26-40 Tache rouge cerise centromaculaire métabolique dans une gangliosidose GM2.
Tableau 26-19 Maladies de surcharge lysosomale pouvant s'associer à une tache rouge ce ¡rise centromaculaire.
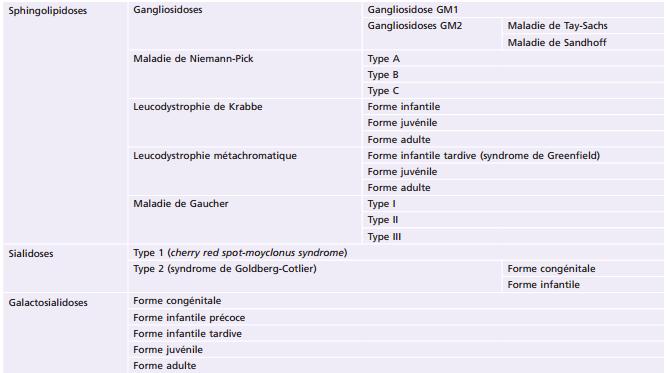
Tableau 26-20 Principales manifestations des sphingolipidoses – les gangliosidoses
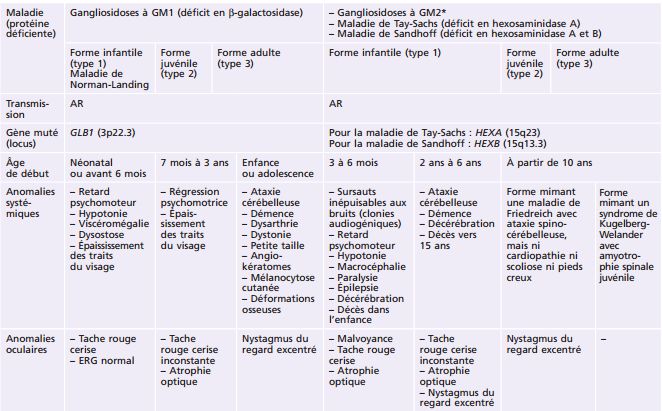
* Par souci de simplification, nous avons choisi de fournir une description unique des manifestations cliniques des gangliosidoses à GM2 d’une part, et de la maladie de Tay-Sachs et de la maladie de Sandhoff d’autre part, car leurs phénotypes se recoupent souvent. Les gènes concernés et les mécanismes de ces deux familles de maladies sont cependant bien distincts. AR : autosomique récessive ; ERG : électrorétinogramme.
Tableau 26-21 Principales manifestations des sphingolipidoses – les maladies de Niemann-Pick et de Gaucher.
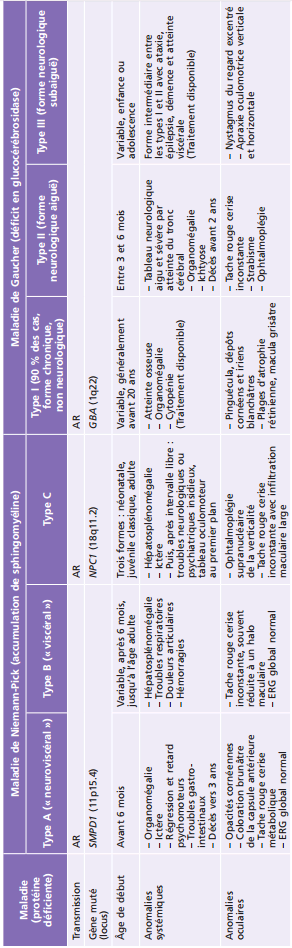
AR : autosomique récessive ; ERG : électrorétinogramme.
Tableau 26-22 Principales manifestations des sphingolipidoses – leucodystrophie de Krabbe et leucodystrophie métachromatique.
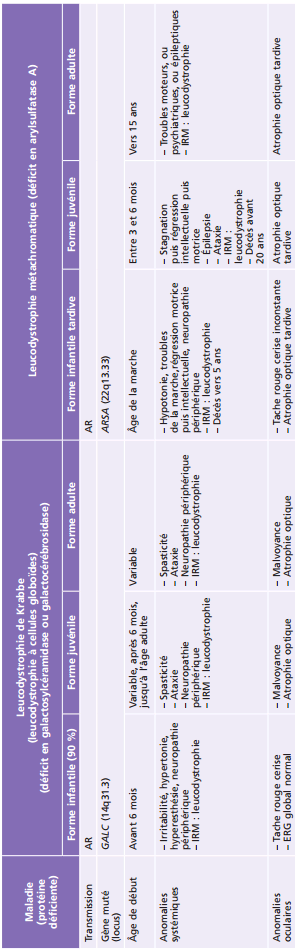
AR : autosomique récessive ; ERG : électrorétinogramme ; IRM : imagerie par résonance magnétique.
Tableau 26-23 Principales manifestations des sialidoses.
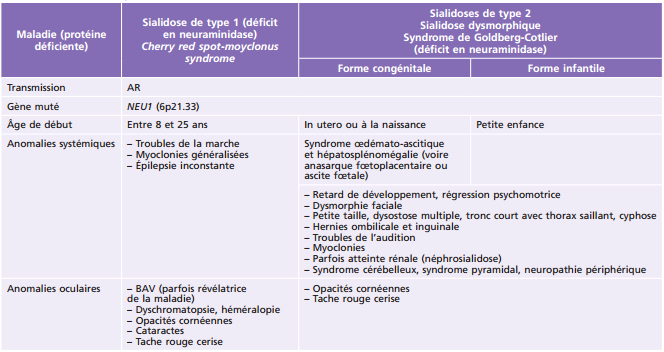
AR : autosomique récessive ; BAV : baisse d’acuité visuelle.
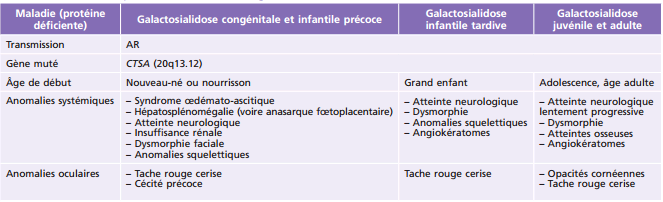
De même que pour les infiltrations cristallines diffuses de la rétine, le diagnostic d'une maculopathie cristalline doit faire éliminer une cause toxique. Tandis que de rares situations évidentes ou idiopathiques sont connues chez l'adulte (drusen calcifiés, télangiectasies juxtafovéales idiopathiques, maculopathie cristalline idiopathique des Igbos du sud-est du Nigéria), les maculopathies cristallines de l'enfant de cause non toxique s'inscrivent dans un cadre syndromique : syndrome de Kjellin (non traité ici en raison de sa physiopathologie encore mystérieuse) et syndrome de Sjögren-Larrson, où l'ophtalmologiste joue un rôle important à l'étape diagnostique. Il faut très soigneusement observer la macula, car les premiers cristaux sont d'abord très fins et difficilement visibles au casque de Schepens (l'usage de l'ophtalmoscope direct nous semble alors essentiel), avant de donner l'aspect classique de maculopathie cristalline typique (fig. 26-41 et tableau 26-25).
Les mucopolysaccharidoses posent deux grands problèmes de prise en charge à l'ophtalmologiste : la problématique de l'infiltration cornéenne et de l'infiltration trabéculaire responsable de glaucome d'une part; la problématique de l'infiltration papillaire et de l'œdème papillaire de stase d'autre part [12]. La papille optique peut être le siège d'une accumulation de glycosaminoglycanes. L'œdème papillaire de stase peut résulter d'une HTIC par hyperpression veineuse cérébrale et/ou d'une infiltration des gaines des nerfs optiques par les glycosaminoglycanes (fig. 26-42). Il est parfois difficile de faire la part entre les différents mécanismes possibles de l'œdème papillaire; les traitements à conduire découlent d'une analyse séméiologique fine multidisciplinaire [25].
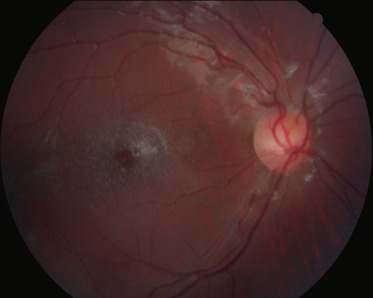
Fig. 26-41 Dystrophie maculaire cristalline dans un syndrome de Sjögren-Larrson.
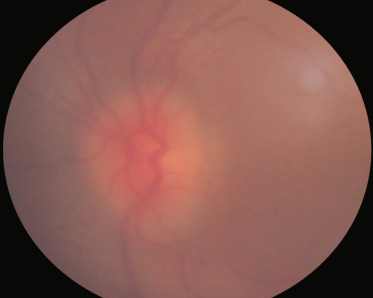
Fig. 26-42 Infiltration de la papille optique par des glycosaminoglycanes dans une mucopolysaccharidose de type 1 (maladie de Hurler).
L’infiltration cornéenne contribue au flou de la photographie.
Tableau 26-25 Principales manifestations du syndrome de Sjögren-Larsson.

AR : autosomique récessive ; ERG : électrorétinogramme.
Les mucopolysaccharidoses sont présentées plus haut dans le cadre des atteintes du segment antérieur.
La maladie de Wilson (MW) est une maladie rare de transmission récessive autosomique associée à des mutations du gène ATP7B (13q14.3) [26, 27]. Sa prévalence est de 0,3/10 000. Elle est due à une anomalie du transport du cuivre du compartiment intracellulaire vers les secteurs extracellulaires, en particulier vers la céru-léoplasmine et la bile. Il en résulte une accumulation de cuivre dans l'organisme (foie, système nerveux central, rein et œil) [27]. L'âge d'apparition dépend des mutations causales. En Europe occidentale, la MW débute tôt, avant l'âge de 10 ans. Elle est à prédominance hépatique et neurologique. Les anomalies oculaires apparaissent dès cet âge. La forme dite « slave » , survenant au-delà de 50 ans, est principalement neurologique. L'accumulation tissulaire du cuivre est responsable du tableau clinique qui associe de façon variable une cirrhose à gros foie, une tubulopathie, une anémie hémolytique, des troubles cardiaques et une atteinte neurologique. Celle-ci se manifeste par un syndrome extrapyramidal avec tremblements, une dysarthrie, des mouvements choréo-athétosiques et une rigidité par lésion des noyaux gris centraux.
Au niveau oculaire, la surcharge en cuivre se manifeste sous forme d'un anneau de couleur cuivrée – la teinte de l'anneau peut être variable – en périphérie de la cornée, l'anneau de Kayser-Fleischer. Souvent incomplet, il est présent chez moins de deux tiers des patients. Il serait cependant constant en cas de manifestations neurologiques. L'anneau apparaît généralement d'abord à midi, puis à 6 heures, puis progresse vers le bas et le haut. Il doit être recherché à la lampe à fente (portable si le patient n'est pas transportable) et ne peut se voir autrement que dans des cas caricaturaux. Il siège au niveau de la membrane de Descemet à la jonction sclérocornéenne et c'est cette zone de midi qu'il faut examiner attentivement. La cataracte caractéristique en fleur de tournesol est rare, de même que l'infiltration choriorétinienne péripapillaire. Les manifestations neuro-ophtalmologiques sont surtout oculomotrices : paralysies de fonction à type de paralysie du regard volontaire vertical puis horizontal, atteinte de la convergence et de l'accommodation et apraxie d'ouverture des paupières.
Tableau 26-26 Principales manifestations des différentes formes de la maladie de Pelizaeus-Merzbacher.
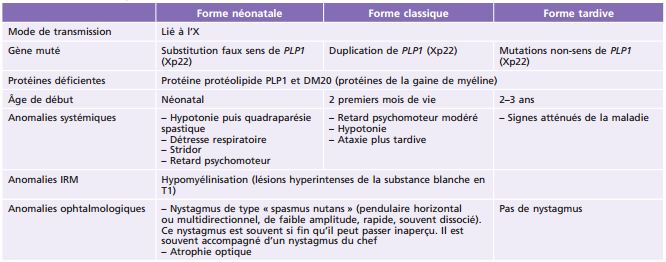
IRM : imagerie par résonance magnétique.
Le traitement de la MW repose sur l'administration de chélateurs du cuivre : sulfate de zinc et D-pénicillamine. La fragmentation et l'atténuation partielle de l'anneau de Kayser-Fleischer constituent un bon test de l'efficacité du traitement. Il peut, dans certaines formes, être nécessaire d'avoir recours à la transplantation hépatique. Enfin, un traitement préventif chez des patients porteurs de mutations à l'état homozygote réduirait la survenue des manifestations symptomatiques [28].
Dans la maladie de Niemann-Piok C, le tableau oculomoteur (parésie puis paralysie oculomotrice de la verticalité, avec atteinte initiale des saccades verticales) est souvent au premier plan et peut amener au diagnostic de la maladie [29]. Les signes de la maladie sont résumés dans le au paragraphe tache rouge cerise métabolique pour des raisons pratiques, bien que celle-ci soit inconstante [24].
MALADIE DE PELIZAEUS-MERZBACHER
La maladie de Pelizaeus-Merzbacher est une leucodystrophie liée à l'X rare. Le nystagmus de type « spasmus nutans » qui s'associe aux autres signes neurologiques de la maladie est évocateur et oriente le diagnostic devant le tableau de leucodystrophie sévère dans la forme néonatale; il peut être le point d'entrée dans la maladie pour la forme classique (tableau 26-26) [30].
[1] Callizo M, Ibanez-Flores N, Laue J Eyelid lesions in lipoid proteinosis or Urbach-Wiethe disease : case report and review of the literature Orbit ( 2011 ) : 30: 242-244
[2] Brady RO, Gal AE, Bradley RM Enzymatic defect in Fabry's disease. Cerami-detrihexosidase deficiency N Engl J Med ( 1967 ) : 276: 1163-1167
[3] Germain DP La maladie de Fabry. Aspects cliniques et genetiques. Perspectives therapeutiques Rev Med Interne ( 2000 ) : 21: 1086-1103
[4] Orssaud C, Dufier J, Germain D Ocular manifestations in Fabry disease : a survey of 32 hemizygous male patients Ophthalmic Genet ( 2003 ) : 24: 129-139
[5] Sher NA, Letson RD, Desnick RJ The ocular manifestations in Fabry's disease Archives of Ophthalmology ( 1979 ) : 97: 671-676
[6] Franceschetti AT Fabry disease : ocular manifestations Birth Defects Orig Artic Ser ( 1976 ) : 12: 195-208
[7] Falke K, Buttner A, Schittkowski M The microstructure of cornea verticillata in Fabry disease and amiodarone-induced keratopathy : a confocal laser-scanning microscopy study Graefes Arch Clin Exp Ophthalmol ( 2009 ) : 247: 523-534
[8] Eng CM, Guffon N, Wilcox WR Safety and efficacy of recombinant human alpha-galactosidase A–replacement therapy in Fabry's disease N Engl J Med ( 2001 ) : 345: 9-16
[9] Al-Hemidan AI, Al-Hazzaa SA Richner-Hanhart syndrome (tyrosinemia type II). Case report and literature review Ophthalmic Genet ( 1995 ) : 16: 21-26
[10] Shams F, Livingstone I, Oladiwura D, Ramaesh K Treatment of corneal cystine crystal accumulation in patients with cystinosis Clin Ophthalmol ( 2014 ) : 8: 2077-2084
[11] Ivanova E, De Leo MG, De Matteis MA, Levtchenko E Cystinosis : clinical presentation, pathogenesis and treatment Pediatr Endocrinol Rev ( 2014 ) : 12: 176-184
[12] Ashworth JL, Biswas S, Wraith E, Lloyd IC Mucopolysaccharidoses and the eye Surv Ophthalmol ( 2006 ) : 51: 1-17
[13] Lindner M, Bertelmann T On the ocular findings in ochronosis : a systematic review of literature BMC Ophthalmol ( 2014 ) : 14: 12 p
[14] Chabrol B, de Lonlay P Maladies métaboliques héréditaires: Doin ( 2011 )
[15] Saudubray JM, Berghe G, Walter JH Inborn metabolic diseases: Springer ( 2012 )
[16] Lloyd IC, Goss-Sampson M, Jeffrey BG Neonatal cataract : aetiology, pathogenesis and management Eye (London, England) ( 1992 ) : 6: 184-196
[17] Endres W, Shin YS Cataract and metabolic disease J Inherit Metab Dis ( 1990 ) : 13: 509-516
[18] Tyni T, Kivela T, Lappi M Ophthalmologic findings in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency caused by the G1528C mutation : a new type of hereditary metabolic chorioretinopathy Ophthalmology ( 1998 ) : 105: 810-824
[19] Fuchs LR, Robert M, Ingster-Moati I Ocular manifestations of cobalamin C type methylmalonic aciduria with homocystinuria J AAPOS ( 2012 ) : 16: 370-375
[20] Small KW, Letson R, Scheinman J Ocular findings in primary hyperoxaluria Archives of Ophthalmology ( 1990 ) : 108: 89-93
[21] Kolker S, Christensen E, Leonard JV Diagnosis and management of glutaric aciduria type I–revised recommendations J Inherit Metab Dis ( 2011 ) : 34: 677-694
[22] Gago LC, Wegner RK, Capone A, Williams GA Intraretinal hemorrhages and chronic subdural effusions : glutaric aciduria type 1 can be mistaken for shaken baby syndrome Retina ( 2003 ) : 23: 724-726
[23] Kafil-Hussain NA, Monavari A, Bowell R Ocular findings in glutaric aciduria type 1 J Pediatr Ophthalmol Strabismus ( 2000 ) : 37: 289-293
[24] Kivlin JD, Sanborn GE, Myers GG The cherry-red spot in Tay-Sachs and other storage diseases Ann Neurol ( 1985 ) : 17: 356-360
[25] Ziyadeh J, Le Merrer M, Robert M Mucopolysaccharidosis type I and craniosynostosis Acta Neurochir (Wien) ( 2013 ) : 155: 1973-1976
[26] Cox DW, Moore SD Copper transporting P-type ATPases and human disease J Bioenerg Biomembr ( 2002 ) : 34: 333-338
[27] Kooy RF, Van der Veen AY, Verlind E Physical localisation of the chromosomal marker D13S31 places the Wilson disease locus at the junction of bands q14.3 and q21.1 of chromosome 13 Human Genet ( 1993 ) : 91: 504-506
[28] Wu ZY, Lin MT, Murong SX, Wang N Molecular diagnosis and prophylactic therapy for presymptomatic Chinese patients with Wilson disease Arch Neurol ( 2003 ) : 60: 737-741
[29] Patterson MC, Hendriksz CJ, Walterfang M Recommendations for the diagnosis and management of Niemann-Pick disease type C : an update Mol Genet Metab ( 2012 ) : 106: 330-344
[30] Hobson GM, Garbern JY Pelizaeus-Merzbacher disease, Pelizaeus-Merzbacher-like disease 1, and related hypomyelinating disorders Semin Neurol ( 2012 ) : 32: 62-67