Pathologie vasculaire
Coordonné par S. Milazzo
S. Milazzo, V. Promelle
La rétinopathie du prématuré (retinopathy of prematurity [ROP]), décrite pour la première fois par Terry en 1942 [1] est une anomalie de la vascularisation rétinienne qui peut conduire lors de son évolution à un handicap visuel majeur, voire à une cécité complète. Elle atteint les nouveau-nés prématurés de faible poids de naissance exposés à une oxygénothérapie.
Sa prise en charge est actuellement bien codifiée et repose sur des classifications et recommandations précises. Les traitements de référence actuels sont la photocoagulation au laser des territoires rétiniens non vascularisés et les injections intravitréennes (IVT) d’agents anti-vascular endothelial growth factor (anti-VEGF).
Les progrès techniques de la réanimation néonatale et la meilleure organisation des soins (régionalisation, travail en réseau) ont conduit à une préservation croissante de nouveau-nés de plus en plus immatures dont le pronostic vital est amélioré. Nos objectifs sont donc de :
– savoir la diagnostiquer et la classer
–savoir la prendre en charge
–connaître le rythme de surveillance.
L’Organisation mondiale de la santé (OMS) a mis en place en 1999 le programme « droit à la vue » pour prévenir les cécités de causes évitables. Le but est d’instaurer des préventions ciblées de ces pathologies, en accord avec des moyens humains et d’infrastructures adaptés (fig. 16-1) [2, 3].
L’incidence actuelle dans les pays développés est évaluée par différentes études.
Aux États-Unis, un registre national (le National Inpatient Sample) a colligé des informations médicales sur toutes les naissances (4,6 millions de naissances vivantes) survenant dans une zone étendue constituant un échantillon représentatif de l’ensemble du pays [4]. Le taux de ROP entre 1997 et 2002 était de 0,12 % chez l’ensemble des enfants nés durant cette période et de 7,34 % chez les prématurés de moins de 37 semaines d’aménorrhée (SA). L’incidence de la ROP était de 12,4 % chez les enfants avec un poids de naissance (PDN) inférieur à 1 250 g.
L’étude américaine ETROP (Early Treatment of Retinopathy Of Prematurity), étude prospective portant sur les indications des traitements des ROP, rapportait une incidence de ROP tous stades confondus de 68 % , chez les enfants de PDN inférieur à 1 251 g entre 2001 et 2002 [5]. Cela est superposable aux résultats de l’étude CRYO-ROP, étude prospective portant sur l’efficacité de la cryothérapie, avec une incidence de 68,5 % sur une population identique entre 1986 et 1987 [6].
Deux grandes études européennes prospectives ont porté sur le devenir des nouveau-nés extrêmes prématurés : l’étude EPIcure au Royaume-Uni et en Irlande ; l’étude EPIBEL en Belgique. L’étude EPIcure a inclus tous les enfants nés entre 20 et 26 SA en 1995 [7] : 811 enfants ont été admis en soins intensifs et 61 % d’entre eux sont décédés avant la sortie. Chez les enfants survivants, 15 % ont bénéficié d’un traitement de la ROP. L’étude EPIBEL a inclus les enfants nés entre 22 et 26 SA entre 1999 et 2000 [8] : 300 enfants prématurés admis en soins intensifs ont été inclus et 175 d’entre eux ont survécu au moment de la sortie de l’hôpital.
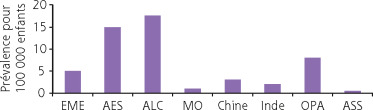
Fig. 16-1 Estimation de la prévalence des déficits visuels sévères et des cas de cécités secondaire à la ROP chez les enfants (nombre pour 100 000 enfants) en fonction du niveau de développement socio-économique.
EME : économies des marchés établis ; AES : anciennes économies socialistes ; ALC : Amérique latine et Caraïbes ; MO : Moyen-Orient ; OPA : autres pays d’Asie ; ASS : Afrique subsaharienne.
Au sein des survivants, le taux de ROP au stade supérieur ou égal à 3 était de 25 % et le taux de ROP au stade seuil était de 20 % .
En France, l’étude EPIPAGE-1 (étude ÉPIdémiologique sur les Petits Âges GEstationnels) a étudié le devenir des nouveau-nés prématurés avant 33 SA ou de PDN inférieur à 1 500 g en 1997. Sur les 2 099 enfants ayant bénéficié d’un examen ophtalmoscopique, 242 présentaient une ROP, soit une incidence de 11,5 % . Sur ces 11,5 % , 9 % étaient de stade 1 ou 2 et 2 % étaient des formes sévères dont seulement trois étaient de stade 4 ; aucun stade 5 n’a été rapporté. L’étude suivante rétrospective EPIPAGE-2, menée sur l’ensemble des enfants nés grands prématurés dans 25 régions de France en 1997, a permis d’inclure 4 290 prématurés et a conduit à constater une amélioration de la survie sans séquelles au-delà de 25 SA [9].
Ces différentes études sont difficilement comparables en raison des différences dans les critères d’inclusions (Tableau 16-1). Toutefois l’incidence de la ROP semble plus élevée aux États-Unis qu’en Europe de l’Ouest. Les dernières études montrent une stabilité dans l’incidence et l’âge d’apparition de la ROP [10].
Les facteurs de risque individualisés sont : le petit poids de naissance inférieur à 1 250 g, la détresse respiratoire et l’hémorragie intraventriculaire. [11].
La vascularisation rétinienne débute in utero au 4e mois de grossesse, à partir de l’artère hyaloïdienne. Au départ du nerf optique, elle progresse en avant vers la périphérie nasale à la 36e semaine, puis vers la périphérie temporale vers la 40e semaine. La prématurité stoppe le développement des vaisseaux rétiniens. La rétine incomplètement vascularisée présente une zone périphérique avasculaire, avec pour conséquence une néovascularisation maximale vers les 32e à 34e SA. Récemment, l’implication des facteurs de croissance a été rapportée pour expliquer les deux phases de la ROP.
Le VEGF, facteur proangiogénique, joue un rôle clé dans le développement vasculaire normal. Le VEGF est indispensable à la survie des cellules endothéliales immatures. La différenciation neuronale provoque une hypoxie physiologique induisant la sécrétion de VEGF par les astrocytes, qui migrent de façon centrifuge de la papille vers la périphérie rétinienne. Les cellules de Müller produisent secondairement du VEGF lors de l’activation des photorécepteurs, activant la formation de la vascularisation rétinienne interne (fig. 16-2). Plusieurs isoformes de VEGF ont été rapportées, mais présentent des fonctions différentes dans le développement vasculaire [12].
La première phase de la ROP évolue de la 22e à la 30e SA :
- l’exposition précoce à des concentrations anormalement hautes d’oxygène entraînerait une hyperoxie relative de la rétine périphérique. Cette hyperoxie induit l’arrêt de la sécrétion du VEGF et donc l’arrêt de la progression des vaisseaux et même la régression des vaisseaux immatures existants. La seconde phase s’installe de la 31e à la 44e SA ;
- l’hypoxie de la rétine ischémique non vascularisée entraîne l’augmentation de la sécrétion de VEGF et d’érythropoïétine (EPO), avec pour conséquence la création de shunts artérioveineux et le développement d’une néovascularisation [13, 14].
D’autres médiateurs, non régulés par l’oxygène, seraient impliqués dans le développement de la ROP, en particulier l’insulinlike growth factor 1 (IGF-1). L’activation du récepteur de l’IGF-1 entraîne l’activation de la sécrétion du VEGF. La chute brutale du taux d’IGF-1 à la naissance, ce dernier provenant du placenta et du liquide amniotique, pourrait jouer un rôle essentiel dans la première phase de la rétinopathie (fig. 16-3).
Voir chapitre 29.
L’International Classification of Retinopathy of Prematurity (ICROP) a publié sa première classification de la ROP en 1984, puis une seconde en 1987. La ROP est classée selon sa localisation par zone, son extension exprimée en quadrants horaires et son acti
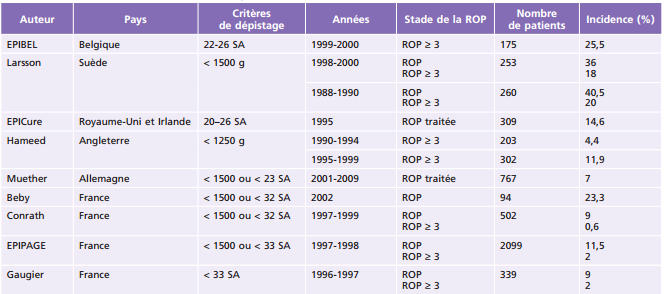
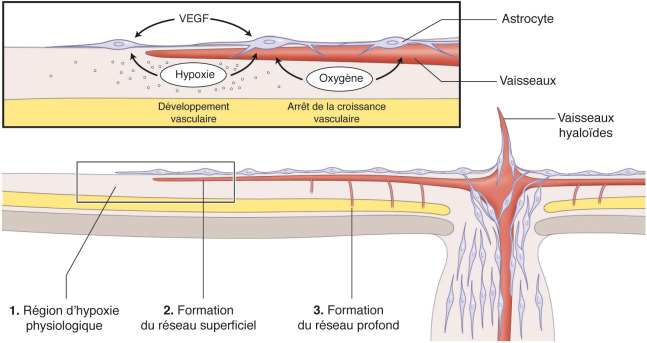
Fig. 16-2 Représentation schématique du rôle de l’oxygène et des astrocytes dans le développement du réseau vasculaire rétinien d’après Saint- Geniez [12].
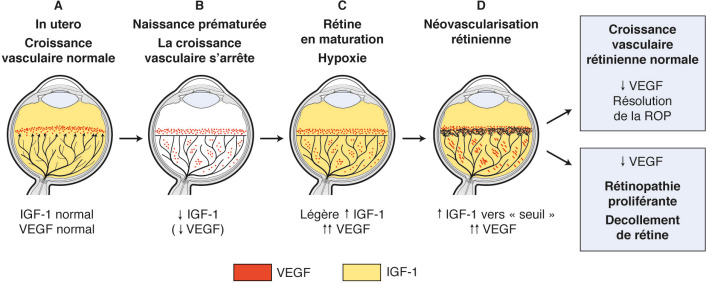
Fig. 16-3 Représentation schématique du développement des vaisseaux sanguins et de son contrôle par le VEGF dans la rétinopathie du prématuré, d’après Smith [13].
vité. Cette classification a été modifiée en 2005 par l’International Committee for the Classification of Retinopathy of Prematurity. Elle apporte deux nouveaux éléments. Le premier est le concept d’une ROP plus virulente, appelée ROP agressive postérieure (aggressive posterior ROP [AP-ROP]). Le second élément est la description d’un stade intermédiaire de dilatation et tortuosité vasculaire portant le nom de pre-plus disease [15].
Le travail photographique du fond d’oeil dans les différents secteurs, plus particulièrement à la RetCam™, représente un moyen d’acquisition et d’archivage pour la surveillance. Elle permet la télétransmission entre pédiatres néonatologistes et ophtalmologistes, évitant ainsi la mobilisation inutile de prématurés aux fonctions vitales souvent fragiles.
La rétine est divisée schématiquement en trois zones concentriques centrées sur la papille (fig. 16-4). Le rayon de la zone 1 (zone centrale) est égal à deux fois la distance inter-papillo-maculaire. La zone 2 démarre à l’extérieur de la zone 1 et atteint l’ora serrata en nasal et l’équateur anatomique en temporal. La zone 3 correspond au croissant résiduel de la rétine antérieure en temporal.
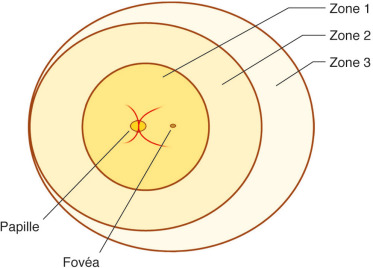
Fig. 16-4 Classification de la rétinopathie du prématuré par localisation en trois zones concentriques centrées sur la papille optique.
L’extension de l’atteinte est précisée par le nombre de quadrants horaires impliqués, et leur caractère contigus ou non. Un quadrant horaire correspond à un secteur de 30°.
En rappel, chez le nouveau-né normal, la jonction entre rétine vascularisée et immature prend l’aspect d’une zone grisée, sans démarcation ou ligne décelable.
La classification en stades repose sur l’aspect jonctionnel rétine vascularisée-rétine avasculaire. Différents stades peuvent être simultanément présents sur le même oeil, l’atteinte est alors cotée sur le stade le plus sévère.
–Stade 1 : il est défini par une ligne de démarcation séparant la rétine avasculaire antérieure de la rétine postérieure vasculaire. Cette fine ligne blanche a un trajet festonné dans le plan rétinien ; des anomalies très polymorphes de ramifications en « brins de balai » peuvent apparaître. Ces derniers peuvent régresser sans séquelles (fig. 16-5a) [15, 16].
- Stade 2 : il se caractérise par un bourrelet rosé surélevé par rapport au plan rétinien. Il se développe à partir de la ligne de démarcation qui s’épaissit en hauteur et en largeur. De petits bouquets néovasculaires peuvent apparaître en arrière du bourrelet. Des vaisseaux pénètrent souvent dans le bourrelet, accentuant son aspect épaissi. Des hémorragies sont possibles (fig. 16-5b) [15, 16].
– Stade 3 : il voit apparaître une néovascularisation et/ou une prolifération fibrovasculaire extrarétinienne s’étendant dans le vitré. Des ponts fibreux (fig. 16-5c), ainsi que des hémorragies sont en général visibles à la base du bourrelet (fig. 16-5d). La sévérité du stade 3 est cotée en minime, modérée ou sévère selon l’extension de la prolifération dans le vitré [15, 16].
– Stades 4 et 5 :
–le stade 4 est le stade de décollement de rétine (DR). Le stade 4A est un DR épargnant la macula et le stade 4B un DR atteignant la zone fovéolaire. L’étendue du DR est cotée comme chez l’adulte. Il s’agit souvent d’un DR tractionnel (fig. 16-5e) [15, 16] ;
– une composante contractile s’ajoute souvent à la traction, aggrave le DR en avant et s’étend en avant jusqu’au cristallin (forme clinique anciennement « fibroplasie rétrolentale » ).
– Stade « pré-plus » : c’est une forme moins marquée de stade « plus » . Ce dernier représente un caractère de gravité et d’évolutivité d’une ROP active. Il se définit par une tortuosité vasculaire au pôle postérieur associée à une dilatation veineuse (fig. 16-5f).
La nomenclature consiste à inclure un symbole « + » au stade identifié, par exemple : stade 2+. Ce stade impose une surveillance étroite, car la ROP peut très rapidement s’aggraver. C’est un véritable marqueur de l’activité de la pathologie.
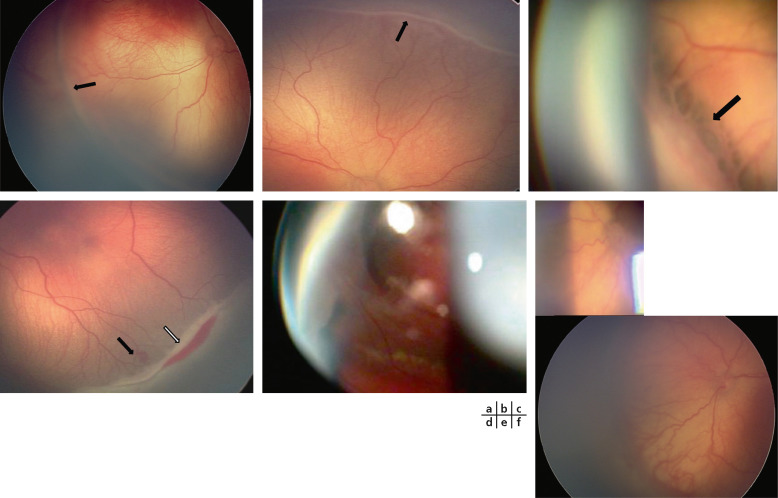
Fig. 16-5 Stades de ROP.
a. Stade 1 : ligne de démarcation entre la rétine vasculaire et avasculaire (cliché à la RetCam™). b. Stade 2 : formation d’un bourrelet au niveau de la ligne de démarcation. c. Stade 3 : ponts fibreux (flèche). d. Stade 3 : hémorragies (flèche pleine) et anomalies vasculaires au pied du bourrelet (flèche vide) (cliché à la RetCam™). e. Stade 4 : décollement de rétine. f. Stade « plus » : dilatations veineuses et artérielles.
La ROP dans sa forme agressive postérieure (AP-ROP), exceptionnelle, ne se voit que chez les grands prématurés. C’est une forme particulièrement sévère qu’il faut identifier et traiter en urgence, au risque d’une évolution foudroyante vers le stade 5. Sa localisation postérieure (en zone 1 le plus souvent), l’importance et l’étendue du stade « plus » associant des dilatations artérielles et veineuses majeures (les artères et les veines devenant difficiles à différencier), l’absence de visualisation de la zone fovéolaire et la formation de shunts entre vaisseaux rétiniens visibles sur l’ensemble de la rétine, y compris sur la rétine vascularisée, en font une entité clinique particulière (fig. 16-6). Un vaisseau circonférentiel peut s’associer à des hémorragies situées à la jonction entre rétine saine et rétine avasculaire. Malgré un traitement bien conduit, le pronostic de ces formes reste encore très réservé.
Ils se définissent depuis 1990 selon l’étude CRYO-ROP [6] :
–le stade seuil (threshold) est une ROP de stade 3+ en zone 1 ou 2 sur au moins cinq secteurs horaires contigus ou huit secteurs horaires cumulatifs ;
–le stade pré-seuil (prethreshold) est une ROP en zone 1 de tout stade ou une ROP en zone 2 de stade 2+ ou 3.
Les paramètres anténatals mis en cause sont l’état septique maternel (chorioamniotite), l’anémie maternelle, le diabète gestationnel, la prise d’antihistaminiques pendant la grossesse, la gémellarité, le retard de croissance intra-utérin, la rupture prolongée des membranes [17].
L’origine ethnique semble impliquée. L’étude CRYO-ROP a observé un taux de ROP stade « seuil » de 7,5 % chez les nouveau- nés d’origine caucasienne contre 3,2 % chez les nouveaunés d’origine afro-américaine [18]. Cette différence selon l’ethnie aurait une origine génétique [19].
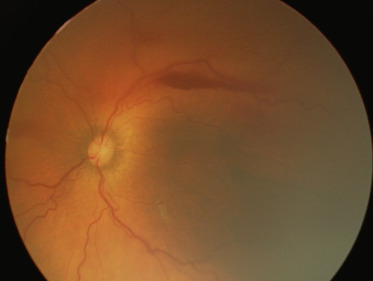
Fig. 16-6 Forme agressive postérieure caractérisée par une absence de zone avasculaire centrale, une tortuosité vasculaire importante.
Apanage des grands prématurés (cliché à la RetCam™).
Les facteurs de risque périnatals et néonatals sont multiples :
- un âge gestationnel bas (inférieur à 30 SA si isolé, ou inférieur à 34 SA si associé à d’autres facteurs de risque) ;
- un PDN faible : pour un PDN compris entre 750 et 1 000 g, le risque de ROP est de 100 % dont 10 % de ROP sévère. Pour un PDN supérieur à 1 500 g, le risque n’est plus que de 1 % [20] ;
- le sexe masculin.
Les critères respiratoires sont importants comme l’existence d’une maladie des membranes hyalines, une ventilation assistée, l’oxygénothérapie prolongée et la dysplasie bronchopulmonaire [20].
– le sexe masculin.
Les critères respiratoires sont importants comme l’existence d’une maladie des membranes hyalines, une ventilation assistée, l’oxygénothérapie prolongée et la dysplasie bronchopulmonaire [20].
Les critères cardiaques et hémodynamiques sont mis en cause comme la persistance du canal artériel et l’utilisation de drogues vasoactives [21, 22].
L’infection est la cause majeure de morbi-mortalité chez les nouveau-nés de PDN inférieur à 1 500 g.
Des critères neurologiques, comme l’hémorragie intraventriculaire, sont étiquetés comme facteurs de risque de la ROP.
Les facteurs de risque sont :
-le poids de naissance inférieur à 1 250 g ;
–la détresse respiratoire ;
- l’hémorragie intraventriculaire [23].
Les atteintes cicatricielles peuvent se rencontrer lors d’une rétinopathie active et évolutive passée inaperçue. Leur présentation est variable selon le degré et la localisation antéropostérieure de l’atteinte :
– myopie avec remaniement pigmentaire périphérique temporal (fig. 16-7a) ;
- traction temporale maculaire, entraînant une pseudo-exotropie par modification de l’angle kappa ;
- fibrose périphérique et contracture des tissus fibreux en un pli rétinien falciforme (fig. 16-7b) ;
- DR tractionnel partiel à total ;
- fibrose rétrolentale, pouvant se compliquer de fermeture de l’angle par déplacement vers l’avant du bloc iridocristallinien.
Son but est de stopper la prolifération anormale des vaisseaux afin de prévenir le DR avec ses conséquences anatomiques et fonctionnelles. Le meilleur traitement est préventif, à savoir la prévention de la prématurité et sa meilleure prise en charge, avec un usage contrôlé de l’oxygénothérapie. La corticothérapie anténatale a été proposée.
Le stade « seuil » (threshold disease) a été défini comme indication de traitement dans l’étude CRYO-ROP [6].
Les indications de traitement ont été élargies en 2003 suite aux résultats de l’étude Early Treatment for Retinopathy of Prematurity [24]. Le traitement est proposé aux rétinopathies « pré-seuil » de type 1, c’est-à-dire :
- en zone 1 quel que soit le stade avec un stade « plus » ;
- en zone 1 de stade 3 sans stade « plus » ;
- en zone 2 avec un stade 2 ou 3 avec stade « plus » .
Tableau 16-2 – Indications du traitement dans les études CRYO-ROP, Early Treatment Study et BEAT-ROP, ayant défini les caractéristiques du stade « seuil » et des rétinopathies « pré-seuil » de type 1 [6, 32].

ROP : retinopathy of prematurity.
Les rétinopathies de type 2 (stade 1 ou 2 sans stade « plus » en zone 1 ou stade 3 sans stade « plus » en zone 2) devront faire l’objet d’une abstention avec surveillance (Tableau 16-2).
La cryothérapie a été le premier traitement proposé dans la ROP dès 1988. La sonde appliquée ab externo délivre du froid qui détruit la rétine périphérique ischémique. L’étude CRYO-ROP a évalué son efficacité : elle a réduit de moitié l’incidence des évolutions défavorables, avec 43 % dans les yeux non traités versus 21,8 % dans les yeux traités [6].
La photocoagulation au laser a supplanté la cryothérapie dans les années 1990, étant source de moins de complications [25–27]. Le laser transpupillaire détruit la rétine ischémique, c’est-à-dire la rétine antérieure au bourrelet. Les impacts doivent être blanc jaunâtre, mais le surdosage doit être évité (fig. 16-8). Ce traitement permet également d’éviter l’angiogenèse floride avant une intervention sur un DR.
La meilleure compréhension de la physiopathogénie a permis de proposer comme solution thérapeutique alternative l’usage des anti-VEGF [14].
Le bévacizumab, injecté seul ou en association au laser, a été le premier étudié. La dilatation pupillaire nécessite fréquemment l’usage de deux collyres mydriatiques : le protocole a déjà été exposé dans un chapitre antérieur [28, 29]. Les IVT ne doivent pas être réalisées dans les stades 4 et 5, au risque de provoquer une contraction des membranes néovasculaires et d’aggraver un DR [30, 31].
En 2011, l’étude pivotale BEAT-ROP [32] compare dans une série prospective, randomisée, les résultats des IVT de bévacizumab versus laser conventionnel dans le traitement des ROP de stade 3+ en zone 1 ou zone 2. Le traitement par bévacizumab intravitréen (0,625 mg dans 0,025 ml de sérum physiologique) s’est révélé plus efficace que le laser pour les ROP de stade 3+ atteignant la zone 1 avec une diminution de 20 % des récidives nécessitant un retraitement. Les deux traitements étaient équivalents pour les ROP n’atteignant pas la zone 1. Le bévacizumab présente l’avantage certain de ne pas détruire la rétine périphérique, permettant à la vascularisation de progresser vers la périphérie (sans toutefois atteindre totalement l’ora) et ainsi de préserver le champ visuel périphérique. L’absence de destruction de la rétine semble d’autant plus intéressante que l’atteinte est postérieure. Toutefois, les récidives, lorsqu’elles surviennent, sont plus tardives après traitement par bévacizumab (16 semaines en moyenne) qu’après laser (6 semaines en moyenne). Cette notion doit être prise en compte par le praticien dans la planification du suivi après traitement. Enfin, le passage systémique des anti-VEGF après IVT est encore en cours d’évaluation. Les données sur la tolérance locale et systémique du VEGF à moyen et long terme sont encore limitées. L’incertitude sur les effets systémiques potentiels à long terme doit être prise en considération dans la décision thérapeutique et expliquée aux parents. Le bévacizumab ne dispose pas d’une autorisation de mise sur le marché (AMM) dans cette indication en France actuellement.
Les formes agressives postérieures répondent mieux au bévacizumab qu’au laser [33].
La dose injectée varie selon les séries. L’efficacité de l’injection de 0,25 mg de bévacizumab dans 0,01 ml de sérum physiologique est identique à une dose de 0,625 mg dans 0,025 ml de sérum physiologique dans les ROP de stade 3 en zone 1 [34]. Le ranibizumab, plus récemment injecté, reste encore en cours d’évaluation.
Lorsque le stade de DR est atteint (stades 4 et 5), une vitrectomie devra le plus souvent être associée au traitement laser. Le pronostic lié à ces stades tardifs est réservé. Lorsque le DR est partiel (stade 4), la chirurgie rétinovitréenne peut permettre une préservation de la macula. En revanche, le DR total (stade 5) résulte en une cécité dans la grande majorité des cas, qu’une vitrectomie ait été réalisée ou non [35, 36]. Cette étape chirurgicale doit être prise en charge par des chirurgiens expérimentés.
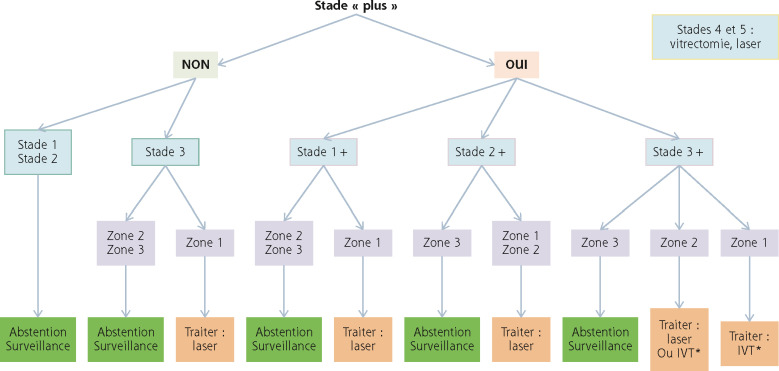
La figure 16-9 résume la prise en charge chirurgicale.
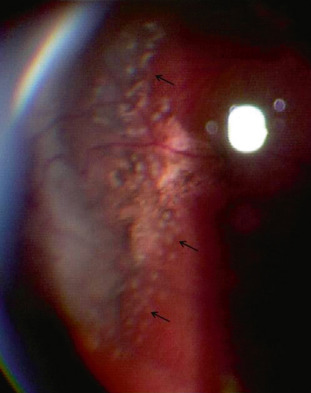
Fig. 16-8 Impacts de laser sur et en arrière du bourrelet.
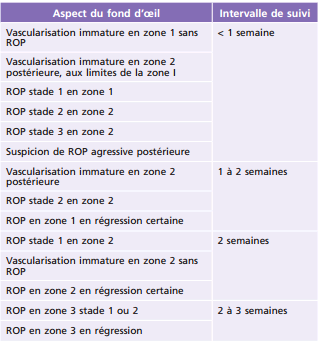
Le rythme de surveillance est résumé dans le Tableau 16-3.
Tableau 16-3 Rythme de surveillance du fond d’oeil en fonction de l’état de la vascularisation de la rétine.
ROP : retinopathy of prematurity. D’après : Fierson WM, American Academy of Pediatrics Section on Ophthalmology, American Academy of Ophthalmology, American Association for Pediatric Ophthalmology and Strabismus, American Association of Certified Orthoptists. Screening examination of premature infants for retinopathy of prematurity. Pediatrics 2013 ; 131 : 189-95.
La ROP évoluerait spontanément vers la régression dans 85 % des cas, toutes formes confondues. Environ 7 % des enfants de PDN inférieur à 1 250 g atteignent le stade « seuil » [16]. L’évolutionsans traitement est alors marquée par le passage progressif à un stade cicatriciel. Celui-ci se caractérise par des degrés variables de fibrose, de contraction des membranes néovasculaires entraînant des tractions vitréorétiniennes, des tractions maculaires et/ou des DR tractionnels.
Les progrès réalisés depuis 1988 sur la compréhension de cette affection et son traitement ont grandement amélioré son pronostic. L’apparition de la cryothérapie puis de la photocoagulation laser a permis de réduire significativement l’incidence de la cécité chez les patients ayant atteint le stade « seuil » [15]. Le traitement par photocoagulation laser d’une ROP en zone 1 serait efficace dans 50 % des cas, permettant de stopper la progression de la rétinopathie. Le traitement précoce des rétinopathies de type 1 permet la réduction du risque d’évolution défavorable sur le plan de l’acuité visuelle de 19,5 à 14,5 % des yeux traités, et sur le plan anatomique de 15,6 à 9 % des yeux traités [37, 38]. Cependant il entraîne inévitablement la perte des cellules rétiniennes périphériques, ce qui est d’autant plus gênant que l’atteinte est postérieure, résultant en une perte significative du champ visuel périphérique et une héméralopie.
La prévention de la ROP est celle de la grande prématurité. Cependant les progrès techniques de la réanimation néonatale permettent à des enfants de plus en plus prématurés de survivre, au prix de soins lourds et de très hautes concentrations en oxygène. Quelques essais ont été réalisés dans l’espoir de diminuer l’incidence et la sévérité de la ROP en modifiant l’exposition à la lumière ou la supplémentation en anti-oxydants sans parvenir à montrer d’efficacité et/ou d’innocuité [39]. L’allaitement maternel pourrait être un facteur protecteur [40]. L’étude SUPPORT, très controversée, a montré que des concentrations en oxygène plus basses (pour une saturation en oxygène comprise entre 85 et 89 % ) pourraient réduire la sévérité de la ROP chez les enfants survivants mais augmenteraient la mortalité en réanimation [41]. Des études sont toujours en cours, évaluant par exemple l’action de molécules permettant le maintien des taux physiologiques d’IGF-1 ou la supplémentation en EPO1. Aucune étude à ce jour ne permet d’identifier avec certitude de véritable facteur de prévention [42].
Certaines atteintes ophtalmologiques se rencontrent plus fréquemment chez les enfants ayant présenté une ROP : myopie, astigmatisme, anisométropie, amblyopie, strabisme, cataracte, glaucome, DR. Certaines sont liées à la prématurité, d’autres aux séquelles de la rétinopathie et de son traitement (en particulier la cryothérapie). Elles peuvent apparaître tardivement, jusque chez le jeune adulte. Un suivi à très long terme s’impose donc chez tous ces patients.
La prématurité s’associerait également à des anomalies maculaires, avec ou sans rétinopathie active : absence de dépression fovéolaire, oedème maculaire cystoïde voire membrane épirétinienne [43].
La ROP est une affection liée à la grande prématurité, survenant chez des enfants aux atteintes multisystémiques. Sa physiopathologie semble aujourd’hui en partie élucidée, ouvrant des options de traitement néanmoins encore limitées. La majorité des ROP, tous stades confondus, évolue favorablement en l’absence de traitement. L’examen systématique du fond d’oeil chez les enfants prématurés à risque de rétinopathie à la RetCam™ permet de dépister les formes nécessitant un traitement et d’améliorer grandement leur pronostic. Il semble que les molécules anti-angiogéniques en IVT ouvrent des perspectives prometteuses, notamment pour les atteintes les plus postérieures. Cependant la faible incidence des formes de ROP nécessitant un traitement, associée à la fragilité du terrain, impose d’évaluer ces traitements dans des études internationales à grande échelle et d’émettre des recommandations de suivi et de prise en charge validées.
[1] Terry TL. Extreme prematurity and fibroblastic overgrowth of persistent vascular sheath behind each cristallinze lens : I. Preliminary report. Am J ophthalmol 1942 ; 25 : 203-4.
[2] Gilbert C, Foster A. Childhood blindness in the context of VISION 2020--the right to sight. Bull World Health Organ 2001 : 79 : 227-32.
[3] Gilbert C, Fielder A, Gordillo L, et al. ; International NO-ROP Group. Characteristics of infants with severe retinopathy of prematurity in countries with low, moderate, and high levels of development : implications for screening programs. Pediatrics 2005 ; 115 : e518-25.
[4] Lad EM, Nguyen TC, Morton JM, Moshfeghi DM. Retinopathy of prematurity in the United States. Br J Ophthalmol 2008 ; 92 : 320-5.
[5] Good WV, Hardy RJ, Dobson V, et al. ; Early Treatment for Retinopathy of Prematurity Cooperative Group. The incidence and course of retinopathy of prematurity : findings from the early treatment for retinopathy of prematurity study. Pediatrics 2005 ; 116 : 15-23.
[6] Palmer EA. Results of U.S. randomized clinical trial of Cryotherapy for ROP (CRYOROP). Doc Ophthalmol 1990 ; 74 : 245-51.
[7] Costeloe K, Hennessy E, Gibson AT, et al. The EPICure study : outcomes to discharge from hospital for infants born at the threshold of viability. Pediatrics 2000 ; 106 : 659-71.
[8] Allegaert K, de Coen K, Devlieger H ; EpiBel Study Group. Threshold retinopathy at threshold of viability : the EpiBel study. Br J Ophthalmol 2004 ; 88 : 239-42
[9] Ancel PY, Goffinet F, EPIPAGE 2 Writing Group, et al. Survival and morbidity of preterm children born at 22 trough 34 weeks’gestation in France in 2011 : results of the EPIPAGE-2 cohort study. JAMA Pediatr 2015 ; 169 : 230-8.
[10] Quinn GE, Barr C, Bremer D, et al. Changes in course of retinopathy of prematurity from 1986 to 2013 : comparison of three studies in the United States. Ophthalmology 2016 ; 12 : S01161-6420.
[11] Lad EM, Nguyen TC, Morton JM, Moshfeghi DM. Retinopathy of prematurity in the United States. Br J Ophthalmol 2008 ; 92 : 320-5.
[12] Saint-Geniez M. Expression et fonction du récepteur MSR/APJ et de son ligand au cours du développement vasculaire de la rétine et de la néovascularisation induite pas l’hypoxie [thèse de doctorat]. Université Paul-Sabatier, Toulouse III ; 2002.
[13] Smith LE. Pathogenesis of retinopathy of prematurity. Semin Neonatol 2003 ; 8 : 469-73.
[14] Smith LE. Through the eyes of a child : understanding retinopathy through ROP the Friedenwald lecture. Invest Ophthalmol Vis Sci 2008 ; 49 : 5177-82.
[15] International Committee for the Classification of Retinopathy of Prematurity. The International Classification of Retinopathy of Prematurity revisited. Arch Ophthalmol 2005 ; 123 : 991-9.
[16] Gilbert C. Retinopathy of prematurity : a global perspective of the epidemics, population of babies at risk and implications for control. Early Hum Dev 2008 ; 84 : 77-82.
[17] Darlow BA, Hutchinson JL, Henderson-Smart DJ, et al. Prenatal risk factors for severe retinopathy of prematurity among very preterm infants of the Australian and New Zealand Neonatal Network. Pediatrics 2005 ; 115 : 990-6.
[18] Saunders RA, Donahue ML, Christmann LM, et al. Racial variation in retinopathy of prematurity. The Cryotherapy for Retinopathy of Prematurity Cooperative Group. Arch Ophthalmol 1997 ; 115 : 604-8.
[19] Lang DM, Blackledge J, Arnold RW. Is Pacific race a retinopathy of prematurity risk factor ? Arch Pediatr Adolesc Med 2005 ; 159 : 771-3.
[20] Beby F, Burillon C, Putet G, Denis P. Retinopathy of prematurity. Results of fundus examination performed in 94 preterm infants. J Fr Ophtalmol 2004 ; 27 : 337-44.
[21] Section on Ophthalmology American Academy of Pediatrics ; American Academy of Ophthalmology ; American Association for Pediatric Ophthalmology and Strabismus. Screening examination of premature infants for retinopathy of prematurity. Pediatrics 2006 ; 117 : 572-6.
[22] Mizoguchi MB, Chu TG, Murphy FM, et al. Dopamine use is an indicator for the development of threshold retinopathy of prematurity. Br J Ophthalmol 1999 ; 83 : 425-8.
[23] Lad EM, Nguyen TC, Morton JM, Moshfeghi DM. Retinopathy of prematurity in the United States. Br J Ophthalmol 2008 ; 92 : 320-5.
[24] Early Treatment For Retinopathy Of Prematurity Cooperative Group. Revised indications for the treatment of retinopathy of prematurity : results of the early treatment for retinopathy of prematurity randomized trial. Arch Ophthalmol 2003 ; 121 : 1684-94.
[25] McNamara JA, Tasman W, Vander JF, Brown GC. Diode laser photocoagulation for retinopathy of prematurity. Preliminary results. Arch Ophthalmol 1992 ; 110 : 1714-6.
[26] Connolly BP, McNamara JA, Sharma S, et al. A comparison of laser photocoagulation with trans-scleral cryotherapy in the treatment of threshold retinopathy of prematurity. Ophthalmology 1998 ; 105 : 1628-31.
[27] Good WV ; Early Treatment for Retinopathy of Prematurity Cooperative Group. Final results of the Early Treatment for Retinopathy of Prematurity (ETROP) randomized trial. Trans Am Ophthalmol Soc 2004 ; 102 : 233-48.
[28] Fierson WM, American Academy of Pediatrics Section on Ophthalmology, American Academy of Ophthalmology, American Association for Pediatric Ophthalmology and Strabismus, American Association of Certified Orthoptists. Screening examination of premature infants for retinopathy of prematurity. Pediatrics 2013 ; 131 : 189-95.
[29] Neffendorf JE, Mota PM, Xue K, Hildebrand GD. Efficacy and safety of phenylephrine 2.5 % with cyclopentolate 0.5 % for retinopathy of prematurity screening in 1246 eye examinations. Eur J Ophthalmol 2015 ; 25 : 249-53.
[30] Honda S, Hirabayashi H, Tsukahara Y, Negi A. Acute contraction of the proliferative membrane after an intravitreal injection of bevacizumab for advanced retinopathy of prematurity. Graefes Arch Clin Exp Ophthalmol 2008 ; 246 : 1061-3.
[31] Zepeda-Romero LC, Liera-Garcia JA, Gutiérrez-Padilla JA, et al. Paradoxical vascular- fibrotic reaction after intravitreal bevacizumab for retinopathy of prematurity. Eye Lond Engl 2010 ; 24 : 931-3.
[32] Mintz-Hittner HA, Kennedy KA, Chuang AZ ; BEAT-ROP Cooperative Group. Efficacy of intravitreal bevacizumab for stage 3+ retinopathy of prematurity. N Engl J Med 2011 ; 364 : 603-15.
[33] Nicoară SD, Ștefănuţ AC, Nascutzy C, et al. Regression rates following the treatment of aggressive posterior retinopathy of prematurity with bevacizumab versus laser : 8-year retrospective analysis. Med Sci Monit 2016 ; 22 : 1192-209.
[34] Han J, Kim SE, Lee SC, Lee CS. Low dose versus conventional dose of intravitreal bevacizumab injection for retinopathy of prematurity : a case series with paired-eye comparison. Acta Ophthalmol 2016 Mar 24.
[35] Repka MX, Tung B, Good WV, et al. Outcome of eyes developing retinal detachment during the Early Treatment for Retinopathy of Prematurity Study (ETROP). Arch Ophthalmol 2006 ; 124 : 24-30.
[36] Caputo G, Metge-Galatoire F, Ardnt C, Conrath J. Rapport de la Société française d’ophtalmologie. Elsevier Masson ; 2011, p. 492-6.
[37] Quinn GE, Barr C, Bremer D, et al. Changes in course of retinopathy of prematurity from 1986 to 2013 : comparison of three studies in the United States. Ophthalmology 2016 ; 123 :1595-600.
[38] Dunbar JA1, Hsu V, Christensen M, et al. Cost-utility analysis of screening and laser treatment of retinopathy of prematurity. J AAPOS 2009 ;13 : 186-90.
[39] Reynolds JD, Hardy RJ, Kennedy KA, et al. Lack of efficacy of light reduction in preventing retinopathy of prematurity. Light Reduction in Retinopathy of Prematurity (LIGHT-ROP) Cooperative Group. N Engl J Med 1998 ; 338 : 1572-6.
[40] Zhou J, Shukla VV, John D, Chen C. Human milk feeding as a protective factor for retinopathy of prematurity : a meta-analysis. Pediatrics 2015 ; 136 : e1576-86.
[41] SUPPORT Study Group of the Eunice Kennedy Shriver NICHD Neonatal Research Network, Carlo WA, Finer NN, et al. Target ranges of oxygen saturation in extremely preterm infants. N Engl J Med 2010 ; 362 : 1959-69.
[42] Fang JL, Sorita A, Carey WA, et al. Interventions To prevent retinopathy of prematurity : a meta-analysis. Pediatrics 2016 ; 137.
[43] Gursoy H, Bilgec MD, Erol N, et al. The macular findings on spectral-domain optical coherence tomography in premature infants with or without retinopathy of prematurity. Int Ophthalmol 2016 ; 36 : 591-600.
S. Milazzo, E. Fauviaux
Les syndromes drépanocytaires sont des pathologies héréditaires responsables de la production anormale d’une ou de plusieurs hémoglobines, entraînant une falciformation des globules rouges. Ils associent trois grandes catégories de manifestations cliniques que sont l’anémie hémolytique chronique, les phénomènes vaso-occlusifs et la susceptibilité aux infections bactériennes. La rétinopathie drépanocytaire, une des complications des syndromes drépanocytaires, se répartit en deux types : la non-proliférante et la proliférante accompagnée de ses potentielles complications comme le DR, l’hémorragie intravitréenne ou le glaucome néovasculaire.
Les syndromes drépanocytaires sont des pathologies héréditaires transmises selon un mode autosomique récessif, secondaires à une anomalie qualitative ou quantitative de l’hémoglobine (Hb). Les mutations de gènes codant pour la structure d’une chaîne polypeptidique (anomalies qualitatives ou drépanocytoses) ou pour une protéine de régulation de la synthèse protéique (anomalies quantitatives ou thalassémies) sont responsables de manifestations cliniques, notamment ophtalmologiques. Ce chapitre étudiera la fréquence élevée des manifestations ophtalmologiques des drépanocytoses chez l’enfant, en opposition aux manifestations ophtalmologiques des thalassémies, qui sont très rares [1].
La drépanocytose est la maladie génétique la plus répandue dans le monde : elle touche plus de 5 millions de personnes à l’état des manifestations cliniques mais selon l’OMS, plus de 120 millions de personnes seraient porteuses d’une mutation drépanocytaire dans le monde. Elle est particulièrement fréquente en Afrique (prévalence pouvant aller jusqu’à 1/30), dans les Antilles (1/250), en Amérique du Nord (États-Unis) et en Amérique du Sud (Brésil). Elle existe également dans les pays du Maghreb, en Sicile, en Grèce et dans tout le Moyen-Orient jusqu’en Arabie saoudite [2]. Sa présence en Europe est due à la migration de populations. En France, la prévalence à la naissance est en moyenne de 1/3000 naissances, mais varie d’une région à l’autre [2] ; 250 nouveaux cas par an sont dépistés en métropole. En raison du dépistage systématique proposé à la naissance, le diagnostic est néonatal.
Cette maladie est due à la mutation du 6e codon de la chaîne bêta-globine, à l’origine de la synthèse d’hémoglobines modifiées : l’hémoglobine S (HbS) et l’hémoglobine C (HbC) étant les plus fréquentes. Les molécules d’hémoglobine drépanocytaire se polymérisent lors de conditions hypoxiques et déforment le globule rouge en lui donnant sa forme caractéristique en faucille (sickle cell). Les hématies, n’étant plus déformables, se bloquent dans les capillaires à l’origine de thromboses.
L’hémoglobine adulte (HbA) normale se compose de quatre chaînes polypeptidiques identiques deux à deux : deux α et deux β branchées sur un noyau hème.
Les hétérozygotes AS portent le trait drépanocytaire, sans manifestation clinique, mais sont transmetteurs. D’autres hémoglobines mutées peuvent s’associer plus rarement à l’HbS telles que les hémoglobines OArab, DPunjab, HbE. Il existe une mutation exceptionnelle appelée SAntilles responsable de manifestations cliniques à l’état hétérozygote. La drépanocytose homozygote SS et les hétérozygoties composites SC, S-thalassémiques, SOArab, SDPunjab, SE sont regroupées sous le terme de syndromes drépanocytaires majeurs. Bien que la forme SS entraîne les manifestations systémiques les plus sévères, ce sont les formes SS et S-thalassémiques qui sont responsables des complications ophtalmologiques graves [2].
Les complications les plus fréquentes de la drépanocytose sont : les crises douloureuses (38,3 % ), la rétinopathie (33,8 % ), la lithiase du cholédoque (30,3 % ), l’ostéonécrose (24,8 % ) et les désordres auditifs (9,7 % ) [1]. Il est clair que si les manifestations vaso-occlusives peuvent toucher tous les territoires, les atteintes oculaires sont très fréquentes et peuvent conduire à la cécité.
Au stade de début, les lésions sont habituellement asymptomatiques. L’examen ophtalmologique systématique est donc obligatoire pour le dépistage.
Il doit être annuel et débuter dès l’âge de 6 ans pour les patients SC et 10 ans pour les patients SS [2]. Les deux facteurs de risque principaux de rétinopathie drépanocytaire sont l’âge et le génotype [2].
Les complications orbitaires dans la drépanocytose sont rares mais un cas récent a décrit la présence d’infarctus orbitaires bilatéraux chez un enfant drépanocytaire de 10 ans, responsables d’un tableau clinique associant exophtalmie, oedème palpébral, ptosis et syndrome de l’apex orbitaire [3]. L’imagerie par résonance magnétique (IRM) a retrouvé la présence d’hématomes intra-orbitaires bilatéraux.
Les signes conjonctivaux sont non spécifiques de la drépanocytose. Il s’agit d’occlusions vasculaires conjonctivales se manifestant par des segments rouge foncé en forme de virgule ou tire-bouchon prédominants au niveau de la conjonctive bulbaire. Ces signes sont plus fréquents dans les formes SS [4].
Au niveau irien, une atrophie segmentaire isolée peut être observée. Une rubéose irienne peut compliquer un glaucome néovasculaire dans les formes proliférantes [4].
L’hyphéma post-traumatique chez un enfant drépanocytaire doit être traité et surveillé de façon rapprochée, car l’hypertonie oculaire est beaucoup plus fréquente que chez le sujet indemne [5]. L’humeur aqueuse favorise la falciformation des hématies du fait de sa pression partielle basse en oxygène et de son pH faible. Ainsi, les hématies falciformées créent de nouvelles falciformations, conduisant à l’hypertonie [6]. La gestion de l’hyphéma posttraumatique et de ses complications est sujette à de nombreuses controverses. Aucune prise en charge n’est standardisée, notamment chez les enfants [7].
La falciformation des hématies entraîne deux phénomènes que sont l’occlusion et la souffrance pariétale des capillaires rétiniens périphériques. La périphérie temporale est le plus souvent touchée. À partir des parois altérées des capillaires se créent des hémorragies rétiniennes, une ischémie chronique, puis des anastomoses artérioveineuses et une néovascularisation par libération de facteurs angiogéniques comme le VEGF et le basic fibroblast growth factor (BFGF) (fig. 16-10). Les occlusions artériolaires se situent surtout au niveau des bifurcations en Y et au niveau des croisements artérioveineux. La topographie des lésions est variable d’un examen à l’autre. Ces lésions s’étendent circonférentiellement et en rétro-équatorial.
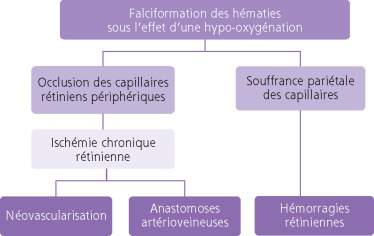
Fig. 16-10 Physiopathologie de la rétinopathie drépanocytaire proliférante (d’après l’EMC syndromes drépanocytaires-physiopathologie de la rétinopathie drépanocytaire).
Cette atteinte peut prendre plusieurs aspects cliniques :
– « blanc sans pression » en plages isolées ou plus allongées ;
– « hémorragies saumonées » qui se situent dans la rétine interne. Elles peuvent être rondes ou ovalaires, à bords bien nets. Leur couleur rouge vif initiale prend vite une teinte saumon en lien avec l’hémolyse progressive (fig. 16-11). Les hémorragies saumonées sont secondaires à la rupture de la barrière hématorétinienne interne (BHRI). La résolution spontanée est la règle. Néanmoins, en cas d’hémorragie abondante, des lésions séquellaires
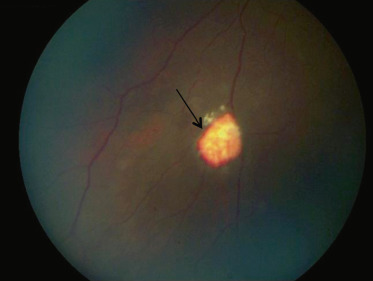
Fig. 16-11 Hémorragie saumonée chez une enfant drépanocytaire de 9 ans.
peuvent être présentes comme le « givre doré » ou les « taches iridescentes » après résorption de l’hémorragie (fig. 16-12) ;
– « black sunburst spots » : ce sont des lésions noires, planes ou arrondies, à contours spiculés, secondaires à l’hyperplasie et à la prolifération focale de l’épithélium pigmentaire induite par des hémorragies. Elles touchent un patient sur deux en moyenne.
La maladie rétinienne au stade clinique s’observe le plus souvent entre 15 et 30 ans et reste donc en général rare chez l’enfant [2]. La progression vers une néovascularisation entraîne un risque d’hémorragie vitréenne et de DR. La cause de néovascularisation est probablement l’occlusion artériolaire au niveau de la périphérie rétinienne, l’ischémie locale résultant de la répétition d’épisodes vaso-occlusifs artériolaires entraînerait une néo-angiogenèse à travers la production de facteurs de croissance vasculaires. Le diagnostic de rétinopathie proliférante nécessite un examen du fond d’oeil suivi d’une angiographie rétinienne permettant une évaluation vasculaire (fig. 16-13).
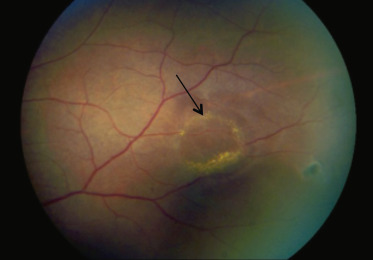
Fig. 16-12 Lésion séquellaire d’une hémorragie saumonée à type de givre doré chez une enfant de 14 ans.
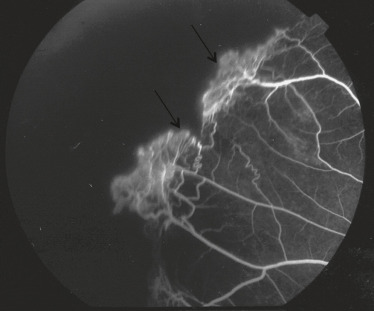
Fig. 16-13 Néovascularisation prérétinienne périphérique en forme de « sea fan » (flèche).
Goldberg propose une classification en cinq stades [8] :
–stade I : occlusions artériolaires périphériques avec un aspect caractéristique en fil d’argent du segment occlus ;
–stade II : anastomose artérioveineuse au niveau de la zone de contact entre rétine perfusée et non perfusée. On ne note pas de diffusion au niveau angiographique, car il s’agit de connexions entre artérioles occluses et veinules terminales adjacentes par le biais de capillaires préexistants dilatés. La barrière hématorétinienne (BHR) est intacte ;
–stade III : néovascularisation prérétinienne périphérique en forme de « sea fan » ou « éventail de mer » . Les néovaisseaux naissent d’une anastomose, puis progressent vers l’avant pour arriver à la jonction vitréorétinienne. On les retrouve surtout en supérotemporal et en inférotemporal, puis moins fréquemment en supéronasal et inféronasal, avec une disposition pré-équatoriale. Ils laissent diffuser la fluorescéine dès les temps précoces avec prise de fluorescéine progressive et intense, signe de rupture de la BHR (fig. 16-13). Ces néovaisseaux peuvent soit régresser spontanément par auto-infarcissement laissant place à des cicatrices fibrogliales, soit proliférer et sont alors à l’origine d’hémorragies intravitréennes et de DR par traction ;
–stade IV : hémorragie intravitréenne. Les néovaisseaux formés peuvent continuer à se développer vers la cavité vitréenne, ce qui peut entraîner un décollement de la hyaloïde postérieure et un risque de saignement du fait des tractions exercées. Lorsque celleci est majeure, le fond d’oeil est inaccessible ;
–stade V : DR. Il est secondaire aux tractions vitréorétiniennes le plus souvent et peut conduire au glaucome néovasculaire. Le diagnostic de rétinopathie proliférante avec néovascularisation est aujourd’hui réalisé à partir de l’angiographie à la fluorescéine. Une étude récente montre que l’angiographie-tomographie par cohérence optique (optical coherence tomography [OCT]) pourrait dépasser le gold standard diagnostique dans la recherche d’ischémie rétinienne chez les enfants drépanocytaires. Un cas de jeune patient avec ischémie rétinienne prouvée à l’angiographie- OCT a été décrit, alors que l’angiographie à la fluorescéine ne retrouvait aucun signe ischémique [9].
La région maculaire peut être atteinte. L’angiographie peut révéler un tableau de maculopathie ischémique, des occlusions artériolaires maculaires pouvant conduire à l’infarctus maculaire. Une étude récente montre que la présence d’une atrophie maculaire temporale (fig. 16-14) est un bon indicateur de présence de néovascularisation périphérique dans la drépanocytose [10]. Les trous maculaires et les membranes épimaculaires peuvent également compliquer une rétinopathie proliférante.
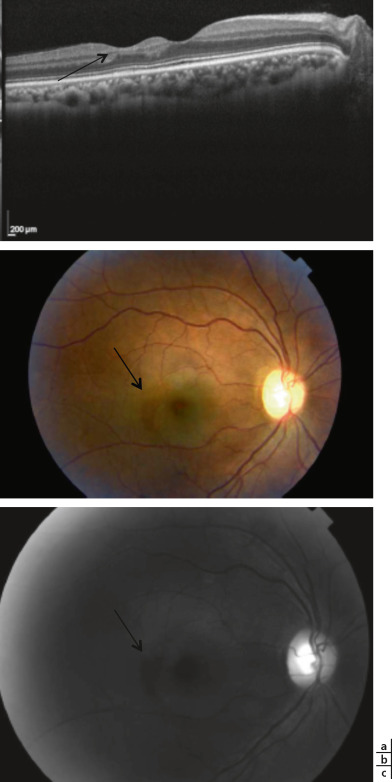
Fig. 16-14 Atrophie maculaire temporale drépanocytaire.
a. Aspect de double macula (flèche) chez une enfant drépanocytaire de 16 ans présentant une atrophie maculaire temporale. b, c. Aspect d’atrophie maculaire temporal (b, flèche) chez une enfant drépanocytaire de 16 ans donnant l’aspect de double macula retrouvée à l’OCT (c, flèche).
L’occlusion de l’artère centrale de la rétine est rare mais existe et peut avoir pour conséquence une atrophie optique. Un cas d’occlusion d’artère centrale de la rétine chez un enfant de 14 ans avec trait drépanocytaire a été décrit récemment [11]. Les stries angioïdes ont également été décrites et peuvent se compliquer de néovaisseaux choroïdiens [12].
Les occlusions vasculaires de la tête du nerf optique sont souvent transitoires et asymptomatiques. Elles apparaissent comme des taches intravasculaires rouge sombre. Il ne faut pas passer à côté du diagnotic de pseudotumor cerebri et savoir rechercher des céphalées sévères et persistantes. Le fond d’oeil montre un oedème papillaire de stase bilatéral. Il est nécessaire d’écarter le diagnostic de thrombose veineuse cérébrale par une imagerie dans cette situation [13].
Lors de la présence de néovaisseaux, il est recommandé de traiter les lésions visibles à l’angiographie par photocoagulation au laser avant d’arriver au stade des complications. D’autres indications peuvent justifier un traitement chirurgical comme l’hémorragie intravitréenne abondante et/ou persistante ou un DR.
La photocoagulation sectorielle confluente est aujourd’hui la méthode de choix dans le traitement des néovaisseaux [14]. Elle diminue la libération des facteurs pro-angiogéniques due à l’ischémie au voisinage des néovaisseaux entraînant leur involution. Les impacts doivent être placés à proximité des sea fans, en avant de ceux-ci, jusqu’à l’ora et sur un secteur le dépassant d’un fuseau horaire de part et d’autre.
La vitrectomie est le traitement des hémorragies intravitréennes importantes et non régressives ou associées à un DR. Elle est réalisée 3 mois après surveillance de l’hémorragie car une résorption spontanée est possible [15]. En cas de chirurgie de DR, des précautions sont indispensables à prendre chez les sujets drépanocytaires. Il faudra : préférer l’anesthésie locorégionale le plus possible ; ne pas utiliser de sympathomimétiques pour la dilatation pupillaire ; éviter l’utilisation de gaz entraînant un risque d’hypertonie oculaire per- et postopératoire ; bannir l’utilisation de Diamox® car celui-ci entraîne un risque d’acidose métabolique ; respecter les quatre muscles droits. La cryo-application et l’indentation doivent être minimalistes. Une des raisons de cette prise de précautions est le risque d’ischémie du segment antérieur. L’éviction des gaz expansifs, la supplémentation en oxygène, le monitorage de la pression intra-oculaire et plus ou moins l’exsanguino-transfusion sont des moyens permettant de lutter contre ce risque [15].
Les recommandations actuelles sont de réaliser un fond d’oeil systématique annuel dès l’âge de 6 ans en cas de drépanocytose de forme SC et dès 10 ans en cas de forme SS.
Cette surveillance sera plus étroite en cas de formes compliquées [16].
D’après l’étude de Rosenberg [17], il faut être encore plus vigilant devant certains antécédents liés à la drépanocytose, comme la présence d’une crise douloureuse ou de séquestration splénique, et un sexe masculin.
Quelques études ont évalué l’efficacité des anti-VEGF et notamment du bévacizumab dans la rétinopathie drépanocytaire proliférante avec des résultats encourageants. Cependant l’innocuité du produit chez l’enfant n’est pas prouvée et d’autres études sont nécessaires pour pouvoir un jour envisager l’utilisation de cette ressource thérapeutique [18]. Par ailleurs, une étude récente a démontré l’efficacité du monométhylfumarate – molécule agissant sur le stress oxydatif et déjà utilisée dans le psoriasis – dans la rétinopathie drépanocytaire chez les souris [19].
[1] Gualandro SF, Fonseca GH, Yokomizo IK, et al. Cohort study of adult patients with haemoglobin SC disease : clinical characteristics and predictors of mortality. Br J Haematol 2015 ; 171 : 631-7.
[2] Benkerrou M, Bernaudin F, Berger C, et al. Syndromes drépanocytaires majeurs de l’enfant et de l’adolescent – Recommandation HAS. HAS ; 2010, p. 47-8.
[3] Schündeln MM, Ringelstein A, Storbeck T, et al. Orbital compression syndrome in a child with sickle cell disease. J Pediatr 2014 ; 164 : 7-9.
[4] Goberville M, Dureau P. Ophtalmologie pédiatrique et strabismes : segment postérieur et neuro-ophtalmologie. Tome 3. Paris : Lavoisier ; 2014 , p. 189-90.
[5] Pandey N. Unusual presentation of ocular trauma in sickle cell trait. Indian J Ophthalmol 2015 ; 63 : 738-40.
[6] Patel AK, Downey L, Dabbs T. Traumatic and postoperative hyphaema in a patient with sickle cell trait. Eye 2004 ; 18 : 212-4.
[7] Bansal S, Gunasekeran DV, Ang B, et al. Controversies in the pathophysiology and management of hyphema, Surv Ophthalmol 2016 ; 61 : 297-308.
[8] Goldberg MF. Classification and pathogenesis of proliferative sickle retinopathy. Am J Ophthalmol 1971 ; 71 : 649-65.
[9] Grover S, Sambhav K, Chalam KV. Capillary nonperfusion by novel technology of OCT angiography in a patient with sickle cell disease with normal fluorescein angiogram. Eur J Ophthalmol 2016 ; 26 : e121-3.
[10] Hood MP, Diaz RI, Sigler EJ, Calzada JI. Temporal macular atrophy as a predictor of neovascularization in sickle cell retinopathy. Ophthalmic Surg Lasers Imaging Retina 2016 ; 47 : 27-34.
[11] Pai SA, Hebri SP, Dekhain MA. Spontaneous central retinal artery occlusion in a teenager with sickle cell trait. Middle East Afr J Ophthalmol 2015 ; 22 : 119-21.
[12] Elagouz M, Jyothi S, Gupta B, Sivaprasad S. Sickle cell disease and the eye : old and new concepts. Surv Ophtalmol 2010 ; 55 : 359-77.
[13] Butori P, Charles A, Elana G, Merle H. Pseudotumor cerebri in children with sickle cell disease. J Fr Ophtalmol 2015 ; 38 : 5-6.
[14] Myint KT, Sahoo S, Thein AW, et al. Laser therapy for retinopathy in sickle cell disease. Cochrane Database Syst Rev 2015 ; 10 : 11-7.
[15] Chen RW, Flynn HW Jr, Lee WH, et al. Vitreoretinal management and surgical outcomes in proliferative sickle retinopathy : a case series. Am J Ophthalmol 2014 ; 157 : 870-5.
[16] HAS. Recommandations pour la pratique clinique : prise en charge de la drépanocytose chez l’enfant et l’adolescent. Septembre 2005. En ligne : http://www.has-sante.fr/ portail/upload/docs/application/pdf/Drepanocytose_reco.pdf
[17] Rosenberg JB, Hutcheson KA. Pediatric sickle cell retinopathy : correlation with clinical factors. J AAPOS 2011 ; 15 : 49-53.
[18] Mitropoulos PG, Chatziralli IP, Parikakis EA, et al. Intravitreal ranibizumab for stage IV proliferative sickle cell retinopathy : a first case report. Case Rep Ophtalmol Med 2014 ; 2014 : 682583.
[19] Promsote W, Powell FL, Veean S, et al. Oral monomethylfumarate therapy ameliorates retinopathy in a humanized mouse model of sickle cell disease. Antioxid Redox Signal 2016 Aug 22.
S. Milazzo, R. Bouvier
La maladie de Coats, aussi appelée télangiectasies rétiniennes primaires, a été décrite pour la première fois en 1908 par Georges Coats [1]. Elle est définie comme une rétinopathie exsudative idiopathique, unilatérale, et touche habituellement des enfants de sexe masculin. Bien que certains cas soient décrits chez l’adulte, il semblerait qu’il existe certaines différences entre populations pédiatrique et adulte [2]. Il n’existe généralement pas d’autre atteinte oculaire ou systémique.
La maladie est caractérisée par la présence au fond d’oeil de télangiectasies capillaires et artériolaires, situées plutôt en périphérie, qui sont responsables d’une exsudation intra- et sous-rétinienne variable pouvant aller jusqu’au DR exsudatif.
Il s’agit d’une pathologie rare dont l’incidence au Royaume-Uni est estimée à 0,09/100 000 habitants [3].
Bien que la physiopathologie ne soit pas connue, l’atteinte vasculaire rétinienne dériverait d’une anomalie de développement de la paroi des capillaires rétiniens.
Après la publication par différents auteurs de nombreux cas atypiques de maladie de Coats, Shields a proposé en 2001 une définition plus précise de la maladie : télangiectasies rétiniennes idiopathiques, associées à une exsudation intra- et sous-rétinienne, sans traction vitréorétinienne détectable [4].
La maladie de Coats est presque exclusivement unilatérale, elle touche les garçons dans 80 % des cas [4] et n’est pas héréditaire. L’âge moyen au moment du diagnostic est d’environ 10 ans, mais des cas sont décrits avant 1 an ou, à l’inverse, au-delà de la cinquantaine [4].
Dans les formes sévères, la maladie est souvent découverte par une leucocorie (fig. 16-15) ou un strabisme par déprivation visuelle. En revanche, les formes modérées et tardives peuvent n’entraîner qu’une simple baisse d’acuité visuelle, ou n’être découvertes que lors d’un examen ophtalmologique de routine.
L’examen du fond d’oeil est primordial. Il permet de mettre en évidence de vastes placards d’exsudats lipidiques intra- et sous-rétiniens, denses et progressant vers la macula (fig. 16-16). Dans cette région, l’évolution se fait volontiers vers la fibrose sous-maculaire. En cas d’exsudation importante, de véritables DR exsudatifs, localisés ou complets, peuvent survenir en l’absence de traitement adapté.
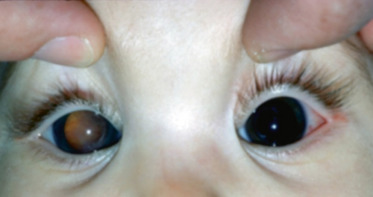
Fig. 16-15 Leucocorie chez un jeune garçon atteint d’une maladie de Coats.
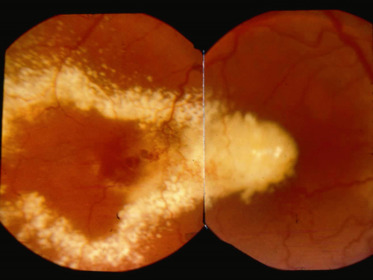
Fig. 16-16 Réaction exsudative dans une maladie de Coats.
C’est l’examen de la périphérie rétinienne qui permet de retrouver les lésions caractéristiques de la maladie de Coats : les télangiectasies. Elles se manifestent par des dilatations anévrismales des artères et des veines sans distinction (fig. 16-17). Ces anévrismes, de forme sacculaire essentiellement, sont localisés préférentiellement dans les quadrants temporal et inférieur de la rétine [5]. En angiographie à la fluorescéine, elles prennent un aspect en ampoules électriques (light bulbs) [4].
Le vitré est généralement clair et rarement le siège d’hémorragie intravitréenne.
En cas de formes évoluées, le segment antérieur peut être le siège d’une rubéose irienne, voire d’un véritable glaucome néovasculaire mettant en péril l’intégrité du globe. La phtyse du globe est la complication ultime de la maladie de Coats.
Même si les manifestations cliniques sont essentiellement unilatérales, des anomalies périphériques à type d’ischémie rétinienne ou d’anévrismes peuvent être retrouvées dans les yeux controlatéraux [6]. Ces anomalies asymptomatiques sont mieux détectées par l’examen angiographique.
Certains signes plus rares peuvent être retrouvés : des shunts artérioveineux ; des micro-anévrismes ; des boucles vasculaires ; des calcifications voire des ossifications compliquant les fibroses sous-maculaires [7] ; des dépôts de cholestérol intravitréens ou en chambre antérieure [8].
Enfin, nous évoquerons simplement les anévrismes miliaires de Leber, qui sont une forme précoce de télangiectasies limitées à la périphérie rétinienne, longtemps asymptomatiques, et de découverte tardive chez l’adulte après 30 ans.
La visualisation chez l’enfant des anévrismes de type Coats en périphérie rétinienne n’est pas toujours aisée et, à l’inverse, une exsudation importante peut les masquer et rendre difficile la différenciation entre Coats et rétinoblastome.
L’imagerie va donc nous aider à affirmer le diagnostic lorsque le doute existe.
L’angiographie à la fluorescéine joue un rôle primordial dans le bilan topographique de l’affection. Elle met en évidence les télangiectasies rétiniennes, sous la forme de capillaires anormalement gros et espacés, d’aspect en ampoules électriques (light bulbs) (fig. 16-18). Elle retrouve aussi d’autres anomalies rétiniennes, comme la dilatation des veinules et des artérioles, des plages d’ischémie (fig. 16-19), des rétinites proliférantes (fig. 16-20) ou des occlusions artériolaires. Elle permet aussi d’apprécier la perméabilité vasculaire, qui fait le pronostic de la maladie, grâce à la diffusion ou non de la fluorescéine [9].
Elle peut également aider en peropératoire à repérer les zones à traiter, grâce aux caméras grand champ type RetCam™ [10].
L’échographie oculaire est un examen simple, nécessitant peu de coopération de l’enfant, et qui permet d’éliminer la présence de masses sous-rétiniennes et de calcifications [11], apanage du rétinoblastome. Rarement, des calcifications peuvent tout de même être rencontrées dans la maladie de Coats.
L’OCT maculaire a un intérêt dans le diagnostic et la surveillance. Il permet de détecter et mesurer un oedème maculaire, et de vérifier l’efficacité d’un traitement sur sa résorption ou non. Il peut parfois retrouver une membrane épirétinienne. Il est à souligner qu’un signe récent a été décrit dans les pathologies maculaires exsudatives, dont la maladie de Coats : le signe du collier de perles (pearl necklace sign) [12], points hyperréflectifs annulaires et contigus à la paroi des espaces cystoïdes de la couche plexiforme externe de la rétine.
La tomodensitométrie contribue à éliminer un rétinoblastome. Comme l’échographie, elle permet d’éliminer la présence de masses sous-rétiniennes ou de calcifications, ou de détecter des anomalies orbitaires ou cérébrales. Il existe néanmoins un risque de faux négatif, notamment pour les rétinoblastomes sans calcifications [13].
Toujours dans le même but, l’IRM présente l’avantage d’être non irradiante et plus sensible que la tomodensitométrie. L’IRM dans la maladie de Coats montre une image hyperintense par rapport au vitré sur les séquences en T1 et T2, non rehaussée par le gadolinium, alors que le rétinoblastome se caractérise à l’IRM par une image hyperintense en T1 mais hypo-intense en T2, et rehaussée par le gadolinium [13].
La cytoponction à l’aiguille fine n’est plus recommandée, car dangereuse et de réalisation difficile.
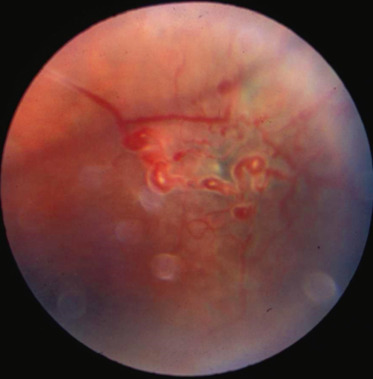
Fig. 16-17 Télangiectasies périphériques typiques de la maladie de Coats.
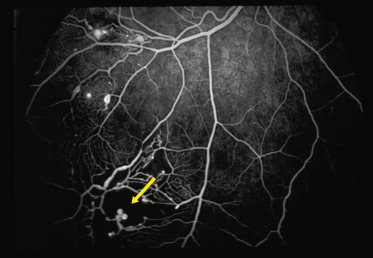
Fig. 16-18 Aspect en ampoules électriques (light bulbs : flèche) des télangiectasies périphériques dans la maladie de Coats.
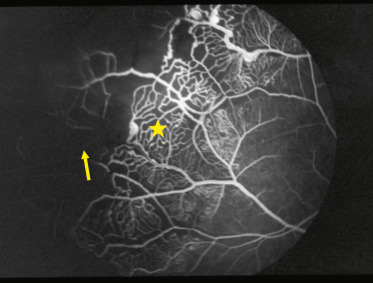
Fig. 16-19 Injection anormale des capillaires rétiniens, zones de non-perfusion (flèche) et aspect typique en échelle (étoile). L’atteinte s’étend jusqu’en périphérie.
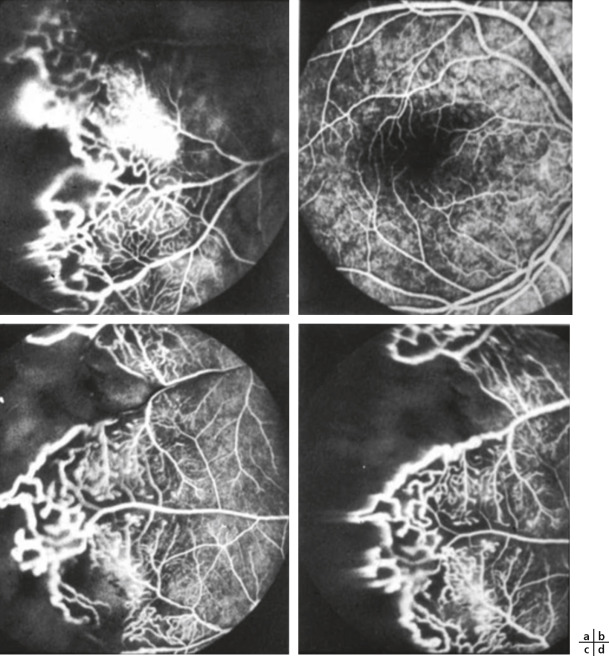
Fig. 16-20 a-d. Rétinites proliférantes bordant des territoires de non-perfusion et anomalies vasculaires typiques de la maladie de Coats.
Actuellement, l’hypothèse physiopathologique concernant la maladie de Coats consiste en une désorganisation de la paroi des capillaires rétiniens, liée à des anomalies de l’endothélium qui devient hyperperméable. La rupture de la BHRI qui en résulte aboutit à une exsudation importante intrarétinienne. Une nécrose des cellules endothéliales pathologiques produirait secondairement les anomalies de la paroi vasculaire à type de dilatation irrégulière, de télangiectasies ou de micro-anévrismes, conduisant à l’ischémie rétinienne.
Les facteurs pro-angiogéniques tels que le VEGF jouent probablement un rôle dans la physiopathologie de la maladie. Une étude portant sur 28 cas de Coats a retrouvé chez ces patients un taux de VEGF intra-oculaire plus important que dans les yeux témoins, ce taux était d’autant plus élevé que le stade de la maladie était avancé [14]. Les injections d’anti-VEGF n’ont fait leurs preuves dans le traitement de la maladie que combinées à d’autres traitements, comme la photocoagulation [15].
Le signe du collier en spectral-domain optical coherence tomography (SD-OCT) a permis d’émettre l’hypothèse que le matériel hyperréflectif est composé de lipoprotéines ou des macrophages chargés de lipides [12].
Bien qu’il n’y ait pas d’hérédité identifiée dans la maladie de Coats, la génétique semble quand même jouer un rôle. Des mutations somatiques acquises du gène NDP, situé sur le chromosome X, ont été retrouvées [16]. Ce gène produit normalement la norrine, protéine qui serait impliquée dans la vasculogenèse normale de la rétine. La prédominance masculine pourrait ainsi s’expliquer par la localisation de ce gène sur le chromosome X. Le gène NDP est également muté dans la maladie de Norrie, qui entraîne une quasiabsence de développement des vaisseaux rétiniens. Yannuzzi [17] a rapporté le cas d’un enfant de 34 mois, présentant des atteintes multisystémiques, dont les tests génétiques étaient positifs au gène « Coats plus » . L’atteinte rétinienne est alors bilatérale.
La maladie de Coats évolue de façon lentement progressive, mais elle se fait globalement vers l’aggravation avec augmentation de l’exsudation lipidique qui conflue vers la macula et décolle progressivement la rétine. Étonnamment, les formes les plus sévères sont fréquemment diagnostiquées chez l’enfant avant 5 ans [3] et conduisent bien souvent à une perte fonctionnelle de l’oeil. En l’absence de traitement adapté, le pronostic anatomique du globe peut être menacé par un glaucome néovasculaire hyperalgique ou une phtyse oculaire.
Dans les cas de Coats moins sévères, le pronostic visuel dépend de l’atteinte maculaire. Les exsudats fovéolaires évoluent volontiers vers la fibrose [9], et ce même après traitement des lésions vasculaires exsudatives.
La classification la plus utilisée est celle proposée par Shields en 2001 [5]. Composée de cinq stades, elle se fonde essentiellement sur l’importance de l’exsudation et permet d’évaluer le potentiel de sauvegarde anatomique du globe (encadré 16-1).
Le stade 1 est caractérisé par la seule présence de télangiectasies limitées en périphérie, sans exsudation. Ce stade asymptomatique est rarement dépisté. Au stade 2, apparaît une exsudation intrarétinienne initialement périphérique autour des lésions vasculaires, épargnant la macula (stade 2A : fig. 16-21a). Puis les exsudats confluent vers le pôle postérieur jusqu’à toucher la fovéola (stade 2B : fig. 16-21b). Au stade 3, on assiste à un DR exsudatif, initialement partiel (stade 3A : fig. 16-21c), puis total (stade 3B : fig. 16-21d) sans traitement adapté. Le stade 4 est marqué par l’apparition d’un glaucome néovasculaire, et le stade 5 correspond au stade terminal de la maladie, avec apparition très souvent d’une phtyse oculaire et d’une cataracte.
Classification de la maladie de Coats d’après Shields et al. [5]
Stade 1 : télangiectasies rétiniennes isolées
Stade 2 : télangiectasies et exsudation :
stade 2A : exsudation extrafovéolaire
stade 2B : exsudation fovéolaire
stade 3A : décollement partiel :
stade 3A1 : extrafovéolaire
stade 3A2 : fovéolaire
stade 3B : décollement total
Stade 3 : décollement de rétine exsudatif :
Stade 4 : décollement de rétine total et glaucome néovasculaire
Stade 5 : stade terminal avec phtyse et cataracte
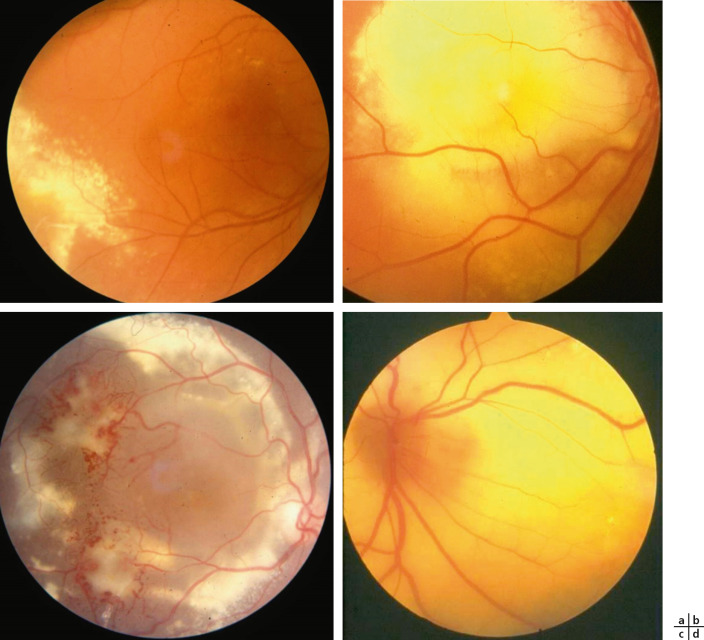
Fig. 16-21 Classification de la maladie de Coats selon Shields.
a. Stade 2A : exsudation périphérique n’atteignant pas la macula. b. Stade 2B : les exsudats confluent jusqu’à la fovéa. c. Stade 3A avec décollement partiel exsudatif. d. Stade 3B avec décollement total.
La grande variabilité des symptômes dans la maladie de Coats peut rendre le diagnostic difficile. En effet, plusieurs pathologies ophtalmologiques peuvent mimer sa présentation clinique. La hantise de tout ophtalmologiste évoquant un Coats doit impérativement et formellement être d’éliminer un rétinoblastome, qui reste le principal diagnostic différentiel du fait de son caractère potentiellement létal.
Le rétinoblastome constitue la tumeur maligne intra-oculaire la plus fréquente chez l’enfant. À la différence de la maladie de Coats, il touche surtout l’enfant en bas âge, sans différence entre les sexes. Dans un contexte familial, l’atteinte est alors souvent bilatérale. Cliniquement, les télangiectasies sont rares dans le rétinoblastome, il n’y a pas d’exsudats lipidiques mais très souvent des calcifications (bien qu’elles puissent rarement se voir dans le Coats). Tous les examens complémentaires décrits précédemment doivent nous aider à éliminer ce diagnostic différentiel. Mais parfois, la différence entre DR exsudatif et rétinoblastome, dans sa forme exophytique essentiellement, ne peut être affirmée avec certitude. L’énucléation reste alors la seule possibilité, en sachant tout de même que le Coats reste la première cause d’énucléation à tort dans ces situations de suspicion de rétinoblastome [4].
D’autres pathologies peuvent évoluer comme la maladie de Coats :
–la rétinopathie du prématuré, dont l’atteinte, fréquemment bilatérale, se situe à l’interface vitréorétinienne plutôt qu’an sein même de la rétine ;
–les hémangiomes capillaires rétiniens de la maladie de von Hippel-Lindau, qui peuvent donner d’importantes exsudations lipidiques, mais dont l’aspect au fond d’oeil ne ressemble pas aux télangiectasies du Coats ;
–la vitréorétinopathie exsudative familiale (VREF) : cette pathologie héréditaire est marquée par l’arrêt du développement des capillaires en périphérie rétinienne. Elle est bilatérale, sans différence entre les sexes, et présente un aspect angiographique et des complications néovasculaires différentes de la maladie de Coats ;
–le rétinoschisis congénital lié à l’X, qui peut exceptionnellement se compliquer de néovascularisation et d’exsudation lipidique importante.
Enfin, nous évoquons simplement certains diagnostics différentiels, tels que l’incontinentia pigmenti, la toxocarose oculaire, la maladie de Eales, la persistance du vitré primitif, les pars planites, etc.
Les objectifs du traitement de la maladie de Coats varient selon l’importance de l’atteinte. Aux stades précoces, le traitement vise à conserver une acuité visuelle fonctionnelle, en détruisant les télangiectasies sources d’exsudation, et à limiter la progression vers un DR. Pour les stades plus avancés, on s’attache avant tout à conserver l’intégrité anatomique du globe.
Pour détruire ces anomalies vasculaires, plusieurs solutions thérapeutiques sont à notre disposition. La photocoagulation au laser reste le traitement de choix dans les stades précoces, lorsque la rétine n’est pas décollée. Les télangiectasies et les zones d’ischémie sont photocoagulées soit à la lampe à fente quand cela est possible, soit au bloc sous anesthésie générale. L’angiographie peropératoire peut avoir un intérêt pour localiser les zones ischémiques [10]. Il est important de traiter toutes les anomalies vasculaires visibles, plusieurs séances sont souvent nécessaires.
Une surveillance régulière avec examen sous anesthésie générale selon l’âge est souvent nécessaire, car il existe un risque de récidive exsudative à partir d’anciennes ou de nouvelles télangiectasies, pouvant apparaître secondairement [5, 13].
Bien qu’il soit admis que le laser ne soit efficace que sur une rétine non décollée, certains auteurs ont trouvé un bénéfice à traiter de façon répétée par photocoagulation des maladies de Coats à des stades avancés (décollements partiels ou subtotaux), et ont réussi à stabiliser la maladie [18, 19].
La cryothérapie garde une place dans l’arsenal thérapeutique. Elle est utilisée dans les mêmes indications que le laser, mais essentiellement lorsque celui-ci est impossible, ou dans les cas de décollements partiels étendus. La cryothérapie peut, comme le laser, être réalisée en plusieurs séances (fig. 16-22).
À partir du stade 3B (décollement total), le laser et la cryothérapie ne sont plus recommandés. Le pronostic fonctionnel est déjà fortement compromis à ce stade et le recours à la chirurgie vitréorétinienne est nécessaire pour éviter l’apparition d’un glaucome néovasculaire. Elle consiste en une vitrectomie, un drainage du liquide sous-rétinien, un tamponnement par gaz ou liquide et un traitement des lésions vasculaires par photocoagulation ou cryothérapie (fig. 16-23).
Aux stades 4 et 5, l’abstention thérapeutique est préférable lorsque l’oeil est indolore. Mais en cas de glaucome néovasculaire sévère ou de phtyse, l’énucléation reste la seule option envisageable.
Récemment, des IVT d’anti-VEGF, notamment le bévacizumab ou le ranibizumab, ont été proposées comme traitement adjuvant dans les maladies de Coats avancées. Ils ont permis, en combinaison avec un traitement standard par photocoagulation ou cryothérapie, une amélioration de l’acuité visuelle ou une préservation de l’intégrité du globe selon les cas [15, 20]. Néanmoins, ce traitement est suspensif et ne dispense pas de la destruction des télangiectasies. Il existerait également un risque de majoration de la fibrose prérétinienne chez les patients traités par anti-VEGF, pouvant aboutir à un DR tractionnel [21]. Ce phénomène serait même accentué en cas de co-traitement par cryothérapie. Enfin, le diagnostic de rétinoblastome doit être formellement éliminé en cas d’intention de traitement par IVT, car il existe un risque de dissémination tumorale. L’évolution post-thérapeutique est fréquemment grevée de complications, telles que des membranes épirétiniennes, sous-rétiniennes, un DR secondaire. Les succès fonctionnels sont rares et modestes.
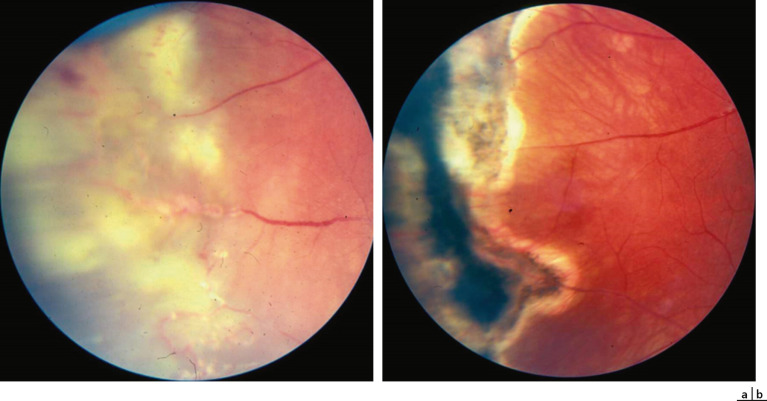
Fig. 16-22 Lésions exsudatives avant (a) et après (b) traitement par cryothérapie.
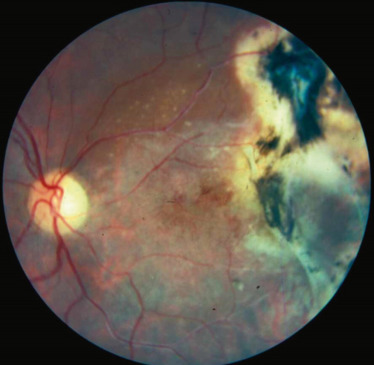
Fig. 16-23 Rétine opérée d’un décollement exsudatif par drainage, tamponnement et cryothérapie périphériques.
Certains auteurs parlent de « syndrome de Coats » pour caractériser des anomalies rétiniennes similaires à celles retrouvées dans la maladie de Coats, associées à divers syndromes et pathologies systémiques. Ces maladies sont listées ci-après de manière non exhaustive :
- rétinite pigmentaire, dont les anomalies vasculaires périphériques pourraient être liées à une mutation du gène CRB1 ;
- dystrophie facio-scapulo-humérale, dont l’atteinte rétinienne est fréquemment bilatérale ;
- syndrome de Turner ;
- syndrome de Cornelia de Lange ;
- syndrome d’Alport ;
- syndrome de Senior-Loken-Schuman ;
- syndrome de leucoencéphalopathie, calcifications et kystes intracérébraux ;
- pseudo-Coats de l’adulte (non détaillé ici).
L’essentiel
La maladie de Coats est une rétinopathie exsudative progressive, unilatérale, touchant essentiellement l’enfant de sexe masculin.
Elle se manifeste chez le petit enfant par un strabisme ou une leucocorie, plus tardivement par une baisse d’acuité visuelle.
L’examen du fond d’oeil retrouve des télangiectasies rétiniennes périphériques typiques de la maladie, des exsudats lipidiques et un DR exsudatif.
L’évolution progressive se fait vers l’aggravation et l’apparition d’un glaucome néovasculaire voire d’une phtyse oculaire.
Il est impératif d’éliminer un rétinoblastome, qui est le principal diagnostic différentiel. Le recours à l’imagerie est nécessaire.
Le traitement comprend la destruction des lésions vasculaires exsudatives par photocoagulation laser ou cryothérapie. La chirurgie vitréorétinienne s’impose en cas de DR important.
Les anti-VEGF doivent être combinés avec un traitement par photocoagulation pour être efficaces.
[1] Coats G Forms of retinal diseases with massive exsudation R Lond Ophthalmol Hosp Rep: ( 1908 ) 440-525
[2] Feng J, Zheng X, Li B, Jiang Y Differences in aqueous concentrations of cytokines in paediatric and adult patients with Coats'disease Acta Ophthalmol: ( 2016 Jun 30 ) -
[3] Morris B, Foot B, Mulvihill A A population-based study of Coats disease in the United Kingdom I : epidemiology and clinical features at diagnosis Eye Lond Engl: ( 2010 ) :24: 1797-1801
[4] Shields JA, Shields CL, Honavar SG, Demirci H Clinical variations and complications of Coats disease in 150 cases : the 2000 Sanford Gifford Memorial Lecture Am J Ophthalmol: ( 2001 ) :131: 561-571
[5] Shields JA, Shields CL, Honavar SG Classification and management of Coats disease : the 2000 Proctor Lecture Am J Ophthalmol: ( 2001 ) :131: 572-583
[6] Blair MP, Ulrich JN, Elizabeth Hartnett M, Shapiro MJ Peripheral retinal nonperfusion in fellow eyes in Coats disease Retina: ( 2013 ) :33: 1694-1699
[7] Miller DM, Benz MS, Murray TG, Dubovy SR Intraretinal calcification and osseous metaplasia in coats disease Arch Ophthalmol: ( 2004 ) :122: 1710-1712
[8] Stacey AW, Borri M, Francesco SD A case of anterior chamber cholestero-losis due to Coats'disease and a review of reported cases Open Ophthalmol J: ( 2016 ) :10: 27-32
[9] Sigler EJ, Calzada JI Retinal angiomatous proliferation with chorioretinal anastomosis in childhood Coats disease : a reappraisal of macular fibrosis using multimodal imaging Retina: ( 2015 ) :35: 537-546
[10] Suzani M, Moore AT Intraoperative fluorescein angiography-guided treatment in children with early Coats'disease Ophthalmology: ( 2015 ) :122: 1195-1202
[11] Atta HR, Watson NJ Echographic diagnosis of advanced Coats'disease Eye Lond Engl: ( 1992 ) :6: 80-85
[12] Gelman SK, Freund KB, Shah VP, Sarraf D The pearl necklace sign : a novel spectral domain optical coherence tomography finding in exudative macular disease Retina: ( 2014 ) :34: 2088-2095
[13] Sigler EJ, Randolph JC, Calzada JI Current management of Coats disease Surv Ophthalmol: ( 2014 ) :59: 30-46 Review
[14] Zhao Q, Peng XY, Chen FH Vascular endothelial growth factor in Coats'disease Acta Ophthalmol (Copenh): ( 2014 ) :92: e22-e28
[15] Villegas VM, Gold AS, Berrocal AM, Murray TG Advanced Coats'disease treated with intravitreal bevacizumab combined with laser vascular ablation Clin Ophthalmol Auckl NZ: ( 2014 ) :8: 973-976
[16] Black GC, Perveen R, Bonshek R Coats'disease of the retina (unilateral retinal telangiectasis) caused by somatic mutation in the NDP gene : a role for norrin in retinal angiogenesis Hum Mol Genet: ( 1999 ) :8: 2031-2035
[17] Yannuzzi NA, Tzu JH, Ko AC Ocular findings and treatment of a young boy with Coats'plus Ophthalmic Surg Lasers Imaging Retina: ( 2014 ) :45: 462-465
[18] Schefler AC, Berrocal AM, Murray TG Advanced Coats'disease. Management with repetitive aggressive laser ablation therapy Retina: ( 2008 ) :28: S38-S41
[19] Shapiro MJ, Chow CC, Karth PA Effects of green diode laser in the treatment of pediatric Coats disease Am J Ophthalmol: ( 2011 ) :151: 725-731
[20] Gaillard MC, Mataftsi A, Balmer A Ranibizumab in the management of advanced Coats disease Stages 3B and 4 : long-term outcomes Retina Phila Pa: ( 2014 ) :34: 2275-2281
[21] Ramasubramanian A, Shields CL Bevacizumab for Coats'disease with exudative retinal detachment and risk of vitreoretinal traction Br J Ophthalmol: ( 2012 ) :96: 356-359
S. Milazzo, O. Khawaja
Pathologie exceptionnelle, la dystrophie facio-scapulo-humérale (DFSH) associe une pathologie musculaire à des anomalies vasculaires rétiniennes périphériques.
La prévalence de la DFSH est de 1 à 5/100 000, ce qui en fait la troisième maladie musculaire génétique rencontrée après la dystrophie de Duchenne et la dystrophie myotonique de type 1 [1]. La maladie commence le plus souvent dans l'enfance ou l'adolescence et touche les deux sexes mais reste d'expression variable puisque moins grave dans la population féminine. Dans de rares cas très précoces, la maladie est plus sévère.
Son mode de transmission est autosomique dominant, mais dans un tiers des cas, il existe une mutation d'apparence sporadique. Le diagnostic repose sur la mise en évidence d'une délétion de 1 à 8 unités répétées, soit 10 à 34 kb [2], dans une séquence d'acide désoxyribonucléique (ADN; D4Z4) au télomère du chromosome 4q (4q35). Cette délétion doit intéresser un allèle de type 4q35 [3].
L'asymétrie et la sélectivité de l'atteinte musculaire caractérisent la DFSH. En effet, seuls certains groupes squelettiques sont touchés, et l'atteinte progresse classiquement de façon lente depuis la partie haute du corps vers les membres inférieurs. La DFSH affecte plus particulièrement les muscles de la face, les fixateurs des omoplates et les muscles du bras, tandis que les muscles oculomoteurs sont épargnés. L'atteinte des muscles de la face se manifeste par un déficit des muscles orbiculaires empêchant l'occlusion complète des paupières. La faiblesse des muscles faciaux supérieurs compromet le recouvrement de la surface oculaire et, en cas de déficit sévère, on peut observer une lagophtalmie et des complications cornéennes à type de kératite. Il est essentiel de prévenir la kératite d'exposition par des larmes artificielles sans conservateur et d'appliquer de la vitamine A au coucher. L'atteinte des muscles de l'épaule est constante. Elle intéresse principalement les muscles fixateurs des omoplates et les muscles élévateurs des bras. Des atteintes extramusculaires sont parfois associées, avec notamment des troubles de l'audition [4], ainsi que des atteintes vasculaires rétiniennes [1].
La DFSH peut donner des atteintes rétiniennes à type de télangiectasies, tortuosités vasculaires et exsudats. L'expression « télangiectasie rétinienne » a été préférée à celle de « maladie de Coats » en raison de la bilatéralité de l'atteinte et de la survenue plus tardive, contrastant avec l'atteinte unilatérale et précoce de la maladie de Coats. La complication la plus sévère associée à cette anomalie vasculaire rétinienne est représentée par une rétinopathie exsudative, identique à celle observée dans la maladie de Coats, qui peut induire des DR exsudatifs récurrents pouvant aboutir à une perte de la vision. Dans des cas plus sévères, une énucléation oculaire peut devenir nécessaire [5].
Une étude incluant 75 patients atteints de DFSH a mis en évidence des télangiectasies des vaisseaux périphériques chez 48 patients sur les 64 qui avaient bénéficié d'une angiographie à la fluorescéine. Seulement 3 d'entre eux présentaient des anomalies des vaisseaux du pôle postérieur associées à une baisse de l'acuité visuelle, tandis que les autres patients étaient totalement asymptomatiques [6]. Dans la majorité des cas, la rétinopathie exsudative est découverte chez les patients pour lesquels un diagnostic de DFSH a déjà été porté, plus rarement décrite comme symptôme initial [5]. Cependant, il a été rapporté un cas de manifestation rétinienne bilatérale avant l'atteinte clinique de DFSH chez un nourrisson de 7 mois qui présentait une tortuosité vasculaire rétinienne associée à des exsudats maculaires et un DR de l'œil droit; la périphérie rétinienne présentait des télangiectasies confirmées par angiographie [7]. Un autre cas a été rapporté chez un enfant de 2 ans qui présentait une surdité sans autre symptôme neurologique ou musculaire. Un examen de son fond d'œil réalisé dans le cadre de l'exploration de sa surdité retrouvait des exsudats et quelques télangiectasies sur la rétine temporale ainsi qu'un décollement séreux rétinien de l'œil gauche. Un examen génétique a confirmé la DFSH avec le portage d'un fragment D4Z4 [8].
Aucune relation ne semble exister entre la sévérité de l'atteinte musculaire et celle de l'atteinte rétinienne, bien que dans la plupart des cas, des rétinopathies exsudatives aient été décrites chez des patients présentant des formes infantiles de dystrophie DFSH [5]. De même, la significativité de l'association entre surdité neurosensorielle et rétinopathie exsudative nécessiterait d'envisager des études plus approfondies. Certaines données de la littérature suggèrent l'existence d'une corrélation entre tortuosité artérielle et sévérité de la DFSH et suggèrent que la tortuosité artérielle pourrait servir de biomarqueur de la sévérité de la DFSH [9].
Sur le plan anatomopathologique, il est intéressant de noter que l'on peut retrouver sur des biopsies musculaires des patients présentant une dystrophie scapulohumérale une inflammation périvasculaire. De même, l'analyse anatomopathologique des globes oculaires énucléés des patients ayant une DFSH montre d'une part, une rétinopathie exsudative majeure avec toutes les caractéristiques de la maladie de Coats et d'autre part, une inflammation périvasculaire chronique de signification inconnue [6]. L'ensemble de ces données a fait émettre l'hypothèse que chez les patients atteints de DFSH, une anomalie primaire endothéliale participerait à l'expression de cette maladie à la fois dans le muscle et dans la rétine. Cette hypothèse est soutenue par une étude récente qui utilise des puces pangénomiques et qui a mis en évidence une altération du profil d'expression de plusieurs gènes ayant un rôle primaire dans le développement des cellules endothéliales et du muscle lisse des vaisseaux sanguins [10].
II est souhaitable que les patients souffrant d'une DFSH bénéficient d'un examen du fond d'œil systématique. Cet examen doit être répété surtout en cas de début précoce de la maladie jusqu'à ce que l'acuité visuelle de l'enfant puisse être évaluée correctement ou chez les patients ayant une DFSH de type 1 porteur d'un fragment D4Z4 [11].
De plus, en cas de doute diagnostique devant une DFSH, l'examen du fond d'œil peut apporter des arguments cliniques en faveur du diagnostic et permettre l'évaluation de la sévérité de la DFSH.
[1] Padberg GW, Brouwer OF, de Keizer RJ, Dijkman G On the significance of retinal vascular disease and hearing loss in facioscapulohumeral muscular dystrophy Muscle Nerve: ( 1995 ) :2: S73-S80
[2] Deidda G, Cacurri S, Piazzo N, Felicetti L Direct detection of 4q35 rearrangements implicated in fascioscapulohumeral muscular dystrophy (FSHD) J Med Genet: ( 1996 ) :33: 361-365
[3] Lemmers RJ, Wohlgemuth M, Frantz RR Contractions of D4Z4 on 4qB subtelomeres do not cause facioscapulohumeral muscular dystrophy Am J Hum Genet: ( 2004 ) :75: 1124-1130
[4] Trevisan CP, Pastorello E, Tomelleri G Facioscapulohumeral muscular dystrophy : hearing loss and other atypical features of patients with large 4q35 deletions Eur J Neurol: ( 2008 ) :15: 1353-1358
[5] Shields CL, Zahler J, Falk N Neovascular glaucoma from advanced Coats disease as the initial manifestation of facioscapulohumeral dystrophy in a 22 years old child Arch Ophtalmol: ( 2007 ) :125: 840-842
[6] Fitzsimons RB, Gurwin EB, Bird AC Retinal vascular abnormalities in facioscapulohumeral muscular dystrophy. A general association with genetic and therapeutic implications Brain: ( 1987 ) :110: 631-648
[7] Ganesh A, Kaliki S, Shields CL Coats-like retinopathy in an infant with preclinical facioscapulohumeral dystrophy J AAPOS: ( 2012 ) :16: 204-206
[8] Lee G, Chen V, Barnes A Retinal telangiectasis detected during a vision screeining examination in a child with hearing loss led to the diagnosis of facioscapulohumeral muscular dystrophy J AAPOS: ( 2014 ) :18: 303-305
[9] Longmuir SQ, Mathews KD, Longmuir RA Retinal arterial but not venous tortusity correlates with facioscaulohumeral dystrophy severity J AAPOS: ( 2010 ) :14: 240-243
[10] Osborne RJ, Welle S, Venance SL Expression profile of FSHD supports a ling between retinal vasculopathy and muscular dystrophy Neurology: ( 2007 ) :68: 569-577
[11] Statland JM, Sacconi S, Farmakidis C Coats syndrome in facioscapulohumeral dystrophy type1 : frequency and D4Z4 contraction size Neurology: ( 2013 ) :80: 1247-1250
S. Milazzo, S. Bryselbout
La rétinopathie diabétique s'observe très rarement chez l'enfant. Le plus jeune patient ayant une rétinopathie diabétique a été diagnostiqué à l'âge de 5 ans et demi; il présentait un microanévrisme [1].
Selon les études, la prévalence de la rétinopathie diabétique chez les enfants est de l'ordre de 9 à 28 % , la majorité ayant une rétinopathie diabétique non proliférante minime à modérée [2]. Dans les travaux de Donaghue et al. [3], 24 % des patients de moins de 18 ans avaient une rétinopathie diabétique après 6 ans d'évolution du diabète de type 1 : ces patients présentaient un taux élevé d'albumine dans le sang.
Le risque de développer une rétinopathie diabétique serait négligeable avant l'âge de 10 ans, même si le diabète est diagnostiqué avant 2 ans d'âge de vie [4]. Chez des patients jeunes, une rétinopathie diabétique proliférante est retrouvée dans 1,2 % des cas après moins de 10 années d'évolution du diabète [5].
Les adolescents ont un risque plus élevé de progression de la rétinopathie diabétique vers la cécité par rapport aux adultes diabétiques.
Des valeurs élevées d'hémoglobine glyquée et de la pression artérielle systolique ou diastolique [6] seraient des facteurs aggravant l'évolution de la rétinopathie diabétique. Tandis que le diagnostic du diabète de type 1 avant l'âge de 5 ans jusqu'à la puberté serait un facteur protecteur de l'apparition de la rétinopathie diabétique [7]. De plus, un équilibre intensif de la glycémie, chez des enfants diabétiques de type 1 de 13 à 17 ans, réduirait le risque de développement d'une rétinopathie diabétique de 53 % [2].
L'adolescence est le meilleur moment pour prendre en charge les facteurs de risque d'évolution vers une rétinopathie diabétique [8] : ces derniers sont résumés dans le Tableau 16-4. La régression de la rétinopathie diabétique est possible chez l'enfant.
Tableau 16-4 – Valeurs cibles des paramètres réduisant le risque de complications microvasculaires chez l’enfant et l’adolescent ayant un diabète de type 1 [8].
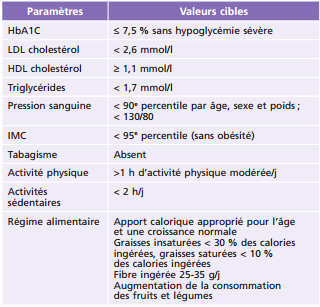
HbA1C : hémoglobine glyquée ; HDH : high density lipoprotein ; IMC : indice de masse corporelle ; LDL : low density lipoprotein.
La rétinopathie diabétique non proliférante est caractérisée par des micro-anévrismes, des hémorragies intra- et prérétiniennes, des exsudats et une tortuosité vasculaire. Elle n'entraîne pas de baisse d'acuité visuelle et n'évolue pas systématiquement vers une rétinopathie proliférante.
La rétinopathie préproliférante est diagnostiquée devant des occlusions vasculaires, des anomalies microvasculaires intrarétiniennes et des nodules cotonneux.
La rétinopathie proliférante est caractérisé par une néovascularisation rétinienne ou de la surface postérieure du vitré. Elle entraîne une baisse d'acuité visuelle si elle se complique d'une hémorragie intravitréenne ou prérétinienne et de DR par traction.
Ces atteintes peuvent être associées à une maculopathie œdémateuse.
Les méthodes de détection les plus sensibles de la rétinopathie diabétique sont la rétinophotographie et l'angiographie à la fluorescéine.
Le traitement de la rétinopathie diabétique par panphotocoagulation rétinienne est indiqué pour des atteintes préprolifératives ou prolifératives et permet de réduire de 50 % la progression vers la cécité.
Il est recommandé de dépister la rétinopathie diabétique par un fond d'œil initial après 3 à 5 ans d'évolution du diabète ou chez des patients de plus de 9 ans [4]. Les enfants atteints de diabète de type 2 ont un risque plus élevé d'avoir une rétinopathie diabétique et doivent être dépistés au moment du diagnostic et dès l'âge de 11 ans [2].
Chaque année, on note une augmentation du risque d'apparition d'une rétinopathie de 16 % chez les enfants diabétiques de type 1 [9].
Complication rare, la papillopathie diabétique survient le plus souvent chez un jeune patient insulino-dépendant.
Il s'agit d'un œdème papillaire transitoire bilatéral qui atteint environ 0,5 % de la population diabétique [10], il se traduit par une baisse d'acuité visuelle, généralement supérieure à 20/50, ou un flou visuel. Les facteurs favorisants le plus souvent retrouvés sont la durée d'évolution du diabète, qui est en moyenne d'une dizaine d'années, et les variations de la glycémie.
Le retentissement fonctionnel est modéré et l'œdème évolue le plus souvent vers une guérison sans séquelles, en l'absence d'une maculopathie associée.
La physiopathologie de cette papillopathie pourrait associer des mécanismes hypoxiques et dysmétaboliques.
La majorité des atteintes ophtalmologiques des syndromes oculo-auditifs avec surdité de perception sont des atteintes rétiniennes. Il existe des syndromes associant surdité de perception et neuropathie optique, le plus connu étant le syndrome de Wolfram.
Le syndrome de Wolfram [11], appelé aussi diabetes insipidus, diabetes mellitus, optic atrophy, and deafness (DIDMOAD), est un syndrome oto-optico-diabétique, dont la prévalence est estimée à 1/700 000. Il s'agit d'un syndrome présentant deux formes cliniques se transmettant sur un mode autosomique récessif.
Le type I est le plus fréquent, il regroupe 90 % des patients. Il se caractérise par l'apparition d'un diabète de type 1 insulino-dépendant se révélant dans l'enfance entre 3 et 10 ans, associé à une atrophie optique bilatérale apparaissant vers 10 ans et aboutissant souvent à la cécité. La surdité, souvent infraclinique dès 12 à 15 ans, ainsi que le diabète insipide, qui se développe plus tard, sont moins fréquents mais constituent, avec le diabète de type 1 et l'atrophie optique bilatérale, la tétrade symptomatologique habituelle. Il existe toutefois une variabilité phénotypique importante et le diagnostic peut être porté sur le seul diabète associé à une atrophie optique. Des anomalies oculaires moins fréquentes incluent des réflexes pupillaires anormaux, un nystagmus, une cataracte, une maculopathie pigmentaire, une rétinopathie (pigmentaire ou diabétique) et un glaucome. D'autres manifestations systémiques sont décrites – troubles neurologiques (ataxie, neuropathies périphériques, vessie neurologique, atonie du tractus urinaire avec hydronéphrose), troubles psychiatriques et démence – qui s'observent également avec une fréquence particulièrement élevée chez les apparentés sains [11].
Le syndrome de Wolfram de type II diffère du précédent par deux points : l'absence de diabète insipide et la présence d'hémorragies digestives secondaires à des ulcères gastriques.
Un gène a été identifié pour chacune des deux formes cliniques :
- –le gène WFS1, localisé sur le chromosome 4 en 4p16.1, codant la wolframine : protéine transmembranaire du réticulum endoplasmique, qui joue un rôle dans l'homéostasie calcique et dans la réponse de la protéine dépliée [12];
- –le gène CISD2, localisé sur le bras long du chromosome 4 en 4q22-24, qui participe au bon fonctionnement mitochondrial. Cette atteinte semble plus fréquente au Moyen-Orient.
Le syndrome de Bardet-Biedl est caractérisé par une rétinopathie pigmentaire de type cone-rod dystrophy, une obésité tronculaire, une polydactylie post-axiale, des anomalies cognitives à type de retard mental, un hypogonadisme chez l'homme, des anomalies génito-urinaires chez la femme et des malformations rénales. Les atteintes secondaires qui s'associent à ce syndrome sont nombreuses : une dysmorphie faciale associée à des anomalies dentaires, un diabète non insulino-dépendant, une hypertension artérielle et une dyslipidémie, des troubles du langage et du développement moteur et psychique, des syndactylies, une ataxie, des anomalies cardiaques et hépatiques, une maladie de Hirschprung, une anosmie. D'autres anomalies ophtalmologiques sont à rechercher : un nystagmus, un strabisme, une forte myopie, une cataracte et un glaucome.
La dégénérescence rétinienne, présente dans 90 % des cas [13], est atypique avec moins de pigmentation que la rétinopathie pigmentaire classique, mais plus sévère avec un électrorétinogramme de très bas voltage ou éteint. Elle évolue vers une cécité précoce, avant l'âge de 20 ans, chez 75 % de ces patients [13].
Le syndrome d'Alstrom est une maladie de système, dont la prévalence est inférieure à 1/100 000 [14], incluant une rétinopathie pigmentaire, une résistance à l'insuline et une hyperinsulinémie, un diabète de type 2 se développant dans l'enfance, une surdité neurosensorielle, une obésité, des atteintes cardiaques, rénales et hépatiques. Le diagnostic associe des critères cliniques et génétiques (Tableau 16-5) [14]. Il se différencie du syndrome de Bardet-Biedl par l'absence de retard mental et d'anomalies digitales. Le diabète non insulino-dépendant, souvent présent dès l'enfance, est accompagné d'atteintes cutanées de type acanthosis nigricans.
La rétinopathie pigmentaire est le symptôme le plus précoce associé à une photophobie et un nystagmus apparaissant entre la naissance et l'âge de 15 mois [14]. Les patients ne ressentent pas d'héméralopie en raison de son évolution rapide. Le fond d'œil retrouve un aspect « poivre et sel » associé à un reflet maculaire cellophanique. Elle évolue vers la cécité dans la deuxième décennie. Un cas de rétinopathie exsudative a été décrit. Le syndrome d'Alstrom est de transmission autosomique récessive à pénétrance variable. Il est dÛ à des mutations du gène ALMS1 sur le chromosome 2 [15].
Les principales cytopathies mitochondriales associant un diabète et une atteinte ophtalmologique sont : le syndrome de MIDD (maternally inherited diabetes and deafness) et le syndrome de MELAS (myopathie mitochondriale, encéphalopathie, acidose lactique). Une maladie mitochondriale doit être évoquée chez des patients présentant une association inexpliquée de symptômes touchant plusieurs organes d'évolution rapidement progressive. La maladie peut avoir un début prénatal, néonatal, dans l'enfance, l'adolescence ou l'âge adulte. Les symptômes ophtalmologiques les plus fréquemment observés sont : une diplopie, un ptosis, une ophtalmoplégie externe progressive, une cataracte, une atrophie optique et une rétinite pigmentaire.
Tableau 16-5 – Critères diagnostiques du syndrome d’Alström [14].
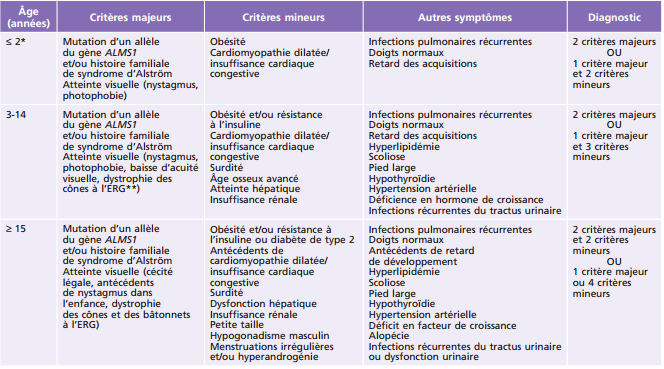
* Ces critères diagnostiques peuvent être réévalués quand le patient est plus âgé.
** L’électrorétinogramme (ERG) doit être réalisé seulement chez des enfants en âge d’effectuer le test.
Le syndrome de MIDD est caractérisé par un diabète et une surdité neurosensorielle de transmission maternelle. Il représente 0,2 à 3 % de tous les cas de diabète. Les patients développent dans plus de 80 % [16] des cas des lésions spécifiques de dystrophie maculaire réticulée, en majorité asymptomatiques. D'autres atteintes, telles que des douleurs musculaires, des troubles gastro-intestinaux, une néphropathie, une cardiomyopathie et des symptômes neuropsychiatriques, sont associées.
L'évolution du syndrome de MIDD est progressive et a un bon pronostic visuel si l'atteinte est localisée à la région périfovéolaire [16].
L'examen du fond d'œil met en évidence des plages d'atrophie choriorétinienne périfovéolaire circonférentielle discontinues [9], mieux visualisées en clichés en autofluorescence et infrarouge [17], et des plages d'hyperpigmentation linéaire autour de la macula et du nerf optique [18].
La rétinopathie diabétique est moins fréquente que chez les patients avec une forme de diabète classique. Le syndrome de MIDD est dÛ dans la majorité des cas à une mutation du gène mitochondrial MT-TL1, qui code l'acide ribonucléique (ARN) mitochondrial de transfert de la leucine, et dans de rares cas à des mutations des gènes mitochondriaux MT-TE et MT-TK qui codent le transfert de l'acide glutamique et de la lysine [16].
Le syndrome de MELAS est une atteinte multisystémique dont les symptômes les plus fréquents sont : des signes neurologiques (céphalées, démence, épilepsie, accidents vasculaires), des signes métaboliques (acidose lactique, diabète), une myopathie et une petite taille. Les atteintes ophtalmologiques incluent une atrophie optique, une rétinopathie pigmentaire et une ophtalmoplégie [19]. Le syndrome de MELAS est dÛ dans la majorité des cas à une mutation du gène mitochondrial MT-TL1.
La rétinopathie est rare chez l'enfant diabétique. L'adolescence est une période potentielle de décompensation.
[1] Holl RW, Lang GE, Grabert M Diabetic retinopathy in pediatric patients with type-1 diabetes : effect of diabetes duration, prepubertal and pubertal onset of diabetes, and metabolic control J Pediatr: ( 1998 ) :132: 790-794
[2] Geloneck MM, Forbes BJ, Shaffer J Ocular complications in children with diabetes mellitus Ophthalmology: ( 2015 ) :122: 2457-2464
[3] Donaghue KC, Craig ME, Chan AK Prevalence of diabetes complications 6 years after diagnosis in an incident cohort of childhood diabetes Diabet Med: ( 2005 ) :22: 711-718
[4] Lueder GT, Pradhan S, White NH Risk of retinopathy in children with type 1 diabetes mellitus before 2 years of age Am J Ophthalmol: ( 2005 ) :140: 930-931
[5] Sultan MB, Sharita C, Huang K Epidemiology, risk factors and management of paediatric diabetic retinopathy Br J Ophtalmol: ( 2012 ) :96: 312-317
[6] Gallego PH, Craig ME, Hing S, Donaghue KC Role of blood pressure in development of early retinopathy in adolescents with type 1 diabetes : prospective cohort study BMJ: ( 2008 ) :337: a918-
[7] Porta M, Schellino F, Montanaro M Prevalence of retinopathy in patients with type 1 diabetes diagnosed before and after puberty Acta Diabetol: ( 2014 ) :51: 1049-1054
[8] Donaghue KC, Chiarelli F, Trotta D Microvascular and macrovascular complications associated with diabetes in children and adolescents Pediatr Diabetes: ( 2009 ) :10: 195-203
[9] Naing A, Kenchaiah M, Krishnan B Maternally inherited diabetes and deafness (MIDD) : diagnosis and management J Diabetes Complications: ( 2014 ) :28: 542-546
[10] Sayin N, Kara N, Pekel G Ocular complications of diabetes mellitus World J Diabetes: ( 2015 ) :6: 92-108
[11] Domenech E, Gomez-Zaera M, Nunes V Wolfram/DIDMOAD syndrome, a heterogenic and molecularly complex neurodegenerative disease Pediatr Endocrinol Rev: ( 2006 ) :3: 249-257
[12] Ribeiro MR, Crispim F, Vendramini MF, Moisés RS Wolfram syndrome : from definition to molecular bases Arq Bras Endocrinol Metabol: ( 2006 ) :50: 839-844
[13] Forsythe E, Beales PL Bardet-Biedl Syndrome Array:University of Washington ( 2003 ) - Seattle 1993–2016. Jul 14 [updated 2015 Apr 23]
[14] Joy T, Cao H, Black G Alstrom syndrome (OMIM 203800) : a case report and literature review Orphanet J Rare Dis: ( 2007 ) :2: 49- Review
[15] Marshall JD, Paisey RB, Carey C, Macdermott S Alström Syndrome Array:University of Washington ( 2003 ) - Seattle 1993–2016. Feb 7 [updated 2012 May 31]
[16] Feigl B, Morris CP Visual function and risk genotypes in maternally inherited diabetes and deafness Can J Ophthalmol: ( 2013 ) :48: e111-e114
[17] Rath PP, Jenkins S, Michaelides M Characterisation of the macular dystrophy in patients with the A3243G mitochondrial DNA point mutation with fundus autofluorescence Br J Ophthalmol: ( 2008 ) :92: 623-629
[18] Massin P, Virally-Monod M, Vialettes B Prevalence of macular pattern dystrophy in maternally inherited diabetes and deafness. GEDIAM Group Ophthalmology: ( 1999 ) :106: 1821-1827
[19] El-Hattab AW, Adesina AM, Jones J3, Scaglia F MELAS syndrome : clinical manifestations, pathogenesis, and treatment options Mol Genet Metab: ( 2015 ) :116: 4-12
S. Milazzo
Bright [1] a décrit pour la première fois en 1836 une insuffisance rénale et des altérations vasculaires du fond de l'œil chez un patient hypertendu.
Chez l'enfant, l'hypertension artérielle (HTA) est le plus souvent asymptomatique. Sa prévalence est de 0,3 à 4,5 % [2]. Son incidence est plus importante si certains facteurs de risque sont associés, comme le surpoids et l'obésité.
La prévalence apparaît augmentée chez les patients en surpoids et souffrant d'obésité : respectivement de 4 à 14 % et de 11 à 23 % [3, 4].
D'autres facteurs de risques sont décrits : régime salé (spécialement chez les enfants obèses et en surpoids), le sexe masculin, l'âge avancé (adolescents versus pré-adolescents) et l'origine ethnique [4].
Chez l'enfant, les normes de pression artérielle sont fonction de l'âge, de la taille et du sexe [5].
Trois grades d'HTA ont été définis en fonction de l'écart par rapport au 95 centile [6] :
- –la pression artérielle est normale lorsque le chiffre de pression systolique et diastolique est inférieur à la valeur du 95 centile;
- –la pression artérielle limite correspond à un écart inférieur à 10 mmHg au-dessus de la valeur du 95 centile : la recherche d'une cause est en général négative. Il s'agit souvent d'enfants avec des facteurs de risque pour une HTA essentielle;
- –l'HTA confirmée correspond à un écart entre 10 et 30 mmHg au-dessus de la valeur du 95 centile : elle doit être systématiquement explorée et traitée;
- –l'HTA menaçante correspond à un écart supérieur à 30 mmHg au-dessus de la valeur du 95 centile : c'est une urgence thérapeutique; les explorations sont conduites de la même manière que pour une HTA confirmée.
Dans tous les cas, un examen ophtalmologique du fond d'œil est obligatoire [5]. Toutefois une étude récente rapporte que cet examen ne devrait pas être systématique, seuls 8,6 % des enfants hypertendus présentant une rétinopathie hypertensive [7]. Une étude récente rétrospective réalisée en réanimation pédiatrique chez des enfants ayant présenté une HTA maligne retrouve des rétinopathies de stade 3 dans 47 % des cas et de stade 4 dans 51 % des cas selon la classification de Keith et Wagener [8].
La découverte d'une rétinopathie hypertensive chez l'enfant doit mener à une enquête étiologique.
L'HTA peut être primitive ou secondaire :
- –primitive, donc sans cause retrouvée, associée aux habitus et aux antécédents familiaux d'HTA;
- –secondaire spécifique d'une maladie principalement rénale, pulmonaire ou médicamenteuse.
Gupta-Malhotra et al. [9] ont colligé dans une étude récente les causes d'HTA chez 423 enfants adressés pour bilan étiologique d'HTA. Sur les 275 enfants présentant réellement une HTA, 156 cas étaient dus à une HTA secondaire versus 119 à une HTA primitive.
Les étiologies les plus fréquentes sont les causes rénales (34 % ), respiratoires (20 % ), médicamenteuses (13 % ) et neurologiques (12 % ).
Les causes rénales sont donc la première étiologie d'HTA secondaire chez l'enfant :
- –glomérulonéphrites aiguës hypertensives;
- –glomérulonéphrites chroniques incluant notamment le syndrome d'Alport, associant une glomérulonéphropathie héréditaire autosomique dominante avec hématurie et surdité de perception;
- –insuffisances rénales aiguës ou chroniques terminales;
- –pathologies rénovasculaires.
Des signes fonctionnels mineurs aspécifiques peuvent être évocateurs : céphalées matinales, en casque, parfois pulsatiles; vomissements inexpliqués; vertiges; crampes; impressions de mouches volantes, de brouillard visuel; bourdonnements d'oreille; paresthésies.
Certaines manifestations témoignent d'une augmentation rapide de la tension artérielle : syndrome polyuropolydipsique; syndrome hémorragique (épistaxis, hémorragie pré- ou postopératoire); cassure de la courbe de croissance staturopondérale; amaigrissement rapide ou encore paralysie faciale récidivante.
Parfois, en cas d'HTA majeure, les manifestations peuvent être bruyantes et nécessiter une prise en charge en urgence : encéphalopathie hypertensive ou œdème aigu pulmonaire.
- –L'interrogatoire des parents est axé sur les antécédents familiaux et personnels de maladies neurologiques, de maladies rénales acquises ou héréditaires, d'une forme héréditaire d'HTA, d'une forme familiale de phéochromocytome, de paragangliome. La triade de Ménard associe céphalées, sueurs et palpitations : elle oriente cliniquement vers le phéochromocytome.
- –L'examen physique recherche : des signes de coarctation de l'aorte, une sténose vasculaire par palpation des pouls, des signes cutanés, des éléments dysmorphiques, des gros reins palpables, des signes d'hyperthyroïdie.
- –Le bilan paraclinique comporte un bilan biologique et une imagerie :
- –bilan biologique : bilan de la fonction rénale (urée sanguine, créatinine plasmatique, protéinurie, dosages de la rénine et de l'aldostérone plasmatiques, dosage des catécholamines urinaires);
- –imagerie :
- –Si le bilan est négatif, la famille sera informée qu'il s'agit d'une HTA essentielle et donc d'un diagnostic d'élimination.
Le tableau clinique de la rétinopathie hypertensive est caractérisé par le rétrécissement artériolaire, les modifications de la paroi des vaisseaux, des altérations du réseau capillaire et les signes cliniques classiques de la rétinopathie : hémorragies, exsudats cotonneux, exsudats lipidiques, atteinte bilatérale (fig. 16-24).

Fig. 16-24 a-c. Enfant de 11 ans atteint d’une leucémie lymphoïde chronique compliquée d’HTA secondaire.
Adressé pour une suspicion de rétinopathie pigmentaire associée à une surdité. L’acuité visuelle est conservée et l’électrorétinogramme est normal. L’examen des rétines montre de larges plages atrophiques périphériques avec remaniement pigmentaire peu marqué (séquelles de taches d’Elschnig). L’interrogatoire retrouve des poussées d’HTA secondaire au traitement par Glivec®. Il s’agit de cicatrices de choroïdopathie hypertensive. L’atteinte est bilatérale.
L'explication physiopathologique est la suivante : l'autorégulation du réseau vasculaire rétinien répond à une augmentation de la pression systémique par une vasoconstriction artérielle. Cela explique les nodules cotonneux et les hémorragies profondes liées à des occlusions artériolaires.
La rupture de la BHR est responsable d'hémorragies rétiniennes superficielles, d'un œdème rétinien et d'exsudats profonds (exsudats secs).
Le premier signe ophtalmoscopique de la rétinopathie hypertensive est la diminution de calibre artériel de façon diffuse ou focale; elle est plus apparente dans sa forme focale. Les modifications de calibre artériel sont plus faciles à apprécier sur des clichés du fond d'œil qu'à l'examen ophtalmoscopique; ils sont réversibles avec le traitement de l'HTA. Mitchell et al. [10] ont mis en évidence, grâce à l'étude de clichés numériques, que le calibre artériel rétinien est inversement corrélé à la pression artérielle, même si la pression artérielle est normale chez les enfants.
Si l'hypertension est sévère ou d'installation rapide, l'autorégulation est dépassée et la BHR est rompue. La rupture de la BHR se traduit par l'apparition d'hémorragies rétiniennes superficielles, un œdème maculaire associé à des exsudats secs (étoile maculaire).
L'œdème papillaire (fig. 16-25), qui s'observe au stade des rétinopathies sévères, est essentiellement la conséquence de troubles circulatoires localisés; il peut être secondaire à une encéphalopathie hypertensive et une augmentation de la pression du liquide céphalorachidien qui doit être éliminée de façon clinique et paraclinique (imagerie cérébrale).
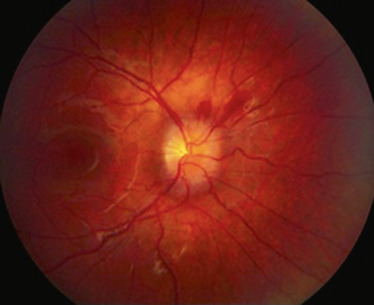
Fig. 16-25 OEdème papillaire entouré d’hémorragies en flammèches lors d’une HTA sévère chez un enfant de 14 ans.
Parmi les différentes classifications, celle de Kirkendall est la plus simple et la plus utilisée :
- –stade I : rétrécissement artériel sévère et disséminé;
- –stade II : en plus des modifications du stade I, présence d'hémorragies rétiniennes, d'exsudats secs et de nodules cotonneux;
- –stade III : en plus des modifications du stade II, présence d'un œdème papillaire.
L'œdème papillaire sera confirmé par angiographie à la fluorescéine.
Les rétrécissements vasculaires liés à l'HTA précèdent les altérations de la paroi vasculaire chez les malades jeunes, mais se développent sur un terrain d'artériosclérose chez les malades âgés. Donc chez les malades hypertendus jeunes, dont les artères au départ sont normales, les modifications de la paroi offrent une estimation de la gravité des altérations et de la durée de l'HTA.
- –Si la poussée hypertensive est brève et prise en charge rapidement, l'acuité visuelle est conservée. Si l'œdème papillaire persiste, on pourra alors observer une baisse d'acuité visuelle et un élargissement de la tache aveugle au champ visuel. Cependant, des complications tardives peuvent apparaître jusqu'à 1 à 2 ans après la poussée : on a décrit des œdèmes maculaires chroniques, des neuropathies optiques progressives, des néovaisseaux choroïdiens [11].
- –Le traitement de la rétinopathie hypertensive est d'abord symptomatique et ensuite étiologique avec prise en charge de l'HTA en urgence. La réduction de la tension artérielle ne doit pas être trop rapide afin d'éviter une ischémie cérébrale ou encore une ischémie aiguë du nerf optique. Il est recommandé une diminution du tiers de l'hypertension sur les 6 premières heures, du deuxième tiers entre 12 et 36 heures, et du dernier tiers entre 48 et 96 heures [12]. Le traitement pédiatrique se heurte à deux problèmes : les médicaments ne font pas l'objet d'une AMM et les posologies et la galénique sont inadaptées à l'utilisation chez l'enfant.
- –Dans les HTA immédiatement menaçantes à prendre en charge en milieu spécialisé, on a recours préférentiellement aux inhibiteurs des canaux calciques (nifédipine ou nicardipine) ou bien aux inhibiteurs de l'enzyme de conversion par voie orale dans un premier temps, si la situation clinique le permet. Si cela reste insuffisant, le recours au furosémide associé à au labétalol injectable est possible. Il faut insister sur la nécessité de la mise en place d'un monitoring tensionnel en ambulatoire [13].
L'HTA chez l'enfant est un problème croissant de santé publique, qui mérite un diagnostic précoce. Certains facteurs de risque, comme l'obésité, sont en hausse, avec leurs conséquences morbides corollaires. Le meilleur traitement reste préventif comportant une activité physique et un régime associé, afin d'éviter un traitement médicamenteux chronique chez ces enfants.
[1] Bright R Tabular view of the morbid appearances in 100 cases connected albuminous urine Guy's Hospital Reports: ( 1836 ) :1: 380-400
[2] Rao G Diagnosis, epidemiology and management of hypertension in children Pediatrics: ( 2016 ) :138: - pii : e20153616
[3] Sorof JM, Lai D, Turner J Overweight, ethnicity, and the prevalence of hypertension in school-aged children Pediatrics: ( 2004 ) :113: 475-482
[4] Freedman DS, Dietz WH, Srinivasan SR, Berenson GS The relation of overweight to cardiovascular risk factors among children and adolescents : the Bogalusa Heart Study Pediatrics: ( 1999 ) :103: 1175-1182
[5] National High Blood Pressure Education Program Working Group on High Blood Pressure in Children and Adolescents. The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents Pediatrics: ( 2004 ) :114: 555-576
[6] Broyer M, Andre JL New look at arterial hypertension in children Arch Fr Pediatr: ( 1980 ) :37: 429-432
[7] Foster BJ, Ali H, Mamber S Prevalence and severity of hypertensive retinopathy in children Clin Pediatr (Phila): ( 2009 ) :48: 926-930
[8] Batouche DD, Kerboua KE, Sadaoui L Clinical and etiological profile malignant hypertension in children in pediatric intensive care Ann Cardiol Angeiol (Paris): ( 2016 ) :65: 165-170
[9] Gupta-Malhotra M, Banker A, Shete S Essential hypertension vs. secondary hypertension among children Am J Hypertens: ( 2015 ) :28: 73-80
[10] Mitchell P, Cheung N, de Haseth K Blood pressure and retinal arteriolar narrowing in children Hypertension: ( 2007 ) :49: 1156-1162
[11] Browning AC, Mengher LS, Gregson RM, Amoaku WM Visual outcome of malignant hypertension in young people Arch Dis Child: ( 2001 ) :85: 401-403
[12] Scharer K Hypertension in children and adolescents Pediatr Nephrol: ( 1987 ) :1: 50-58
[13] Flynn JT, Daniels SR, Hayman LL Update : ambulatory blood pressure monitoring in children and adolescents : a scientific statement from the American Heart Association Hypertension: ( 2014 ) :63: 1116-1135
S. Milazzo, M. Badguerahanian
La maladie de Eales est une vascularite rétinienne périphérique idiopathique. Elle doit son nom à un ophtalmologiste Henry Eales, qui a décrit des tableaux d'hémorragies intravitréennes récidivantes chez de jeunes adultes âgés de 14 à 29 ans en 1980 [1]. Wardsworth apporta des précisions, quelques années plus tard, en décrivant des signes inflammatoires associés [2].
On compte la plupart des cas décrits en Inde, où l'incidence est de 1 patient sur 250 [3]. Cette pathologie touche principalement les hommes et se développe entre 20 et 30 ans [4]. Quelques cas ont été décrits chez l'enfant de moins de 18 ans; 33 yeux sur 82 (40 % ) présentant une hémorragie intravitréenne spontanée étaient atteints par une maladie de Eales dans une étude indienne de Rishi et al. [5]. En revanche, Spirn et al. [6] ne rapportent qu'un cas de maladie de Eales sur 186 hémorragies intravitréennes chez les moins de 18 ans, en excluant les rétinopathies du prématuré actives, mais avec une moyenne d'âge plus faible que dans l'étude de Rishi.
Bien que d'étiologie inconnue, plusieurs hypothèses ont été émises. La première hypothèse est celle d'un mécanisme autoimmun (exposition à un agent extérieur entraînant une libération d'un antigène uvéitopathogénique provoquant une réponse immune intra-oculaire à l'origine de la maladie) [7]. La deuxième hypothèse est celle d'une implication d'une réponse immune médiée par les cellules T [8]. La troisième hypothèse est celle d'une association à la tuberculose, une étude retrouvant la présence du génome de Mycobacterium tuberculosis dans 57 % des vitrés des patients atteints et vitrectomisés dans le cadre de maladie de Eales [9, 10 ]. Une recherche de tuberculose doit être pratiquée en cas de suspicion de maladie de Eales par la réalisation d'une intradermoréaction à la tuberculine et d'un quantiféron.
La maladie est caractérisée par la triade inflammation (vascularite rétinienne périphérique)-modifications ischémiques (non-perfusion rétinienne périphérique)-néovascularisation de la rétine et du nerf optique qui entraîne souvent une hémorragie intravitréenne, mode de révélation habituel de la pathologie.
Les symptômes liés à l'hémorragie intravitréenne ne survenant qu'après la néovascularisation, les enfants sont souvent asymptomatiques au stade initial de la maladie. Les plaintes initiales aspécifiques sont liées à l'hémorragie intravitréenne : baisse d'acuité visuelle variable allant de « voit bouger la main » à une acuité visuelle conservée, myodésopsies, flou visuel, de façon unilatérale. Cinquante à 90 % des patients présenteront une bilatéralisation de l'atteinte [11, 12 ].
Les signes sont retrouvés en biomicroscopie :
- –l'uvéite antérieure de façon très peu fréquente : 3 cas sur 466 d'après Atmaca [12];
- –la vascularite de topographie périphérique (fig. 16-26) : exsudats le long des veines rétiniennes, hémorragies rétiniennes superficielles, engainements veineux rétiniens;
- –une occlusion de branche veineuse rétinienne périphérique dans 3,2 % des cas associés aux signes de vascularites dans un autre quadrant [13];
- –une hémorragie intravitréenne dans un tiers des cas [12];
- –une néovacularisation rétinienne au niveau de la jonction rétine perfusée-non perfusée;
- –une néovascularisation du nerf optique;
- –un DR tractionnel, retrouvé dans 2,5 à 13 % des cas [12, 14 ].
- –un œdème maculaire, des exsudats maculaires ou des membranes épirétiniennes constituant une évolution possible, mais rare. La présence d'une choriorétinite doit faire rechercher un autre diagnostic.
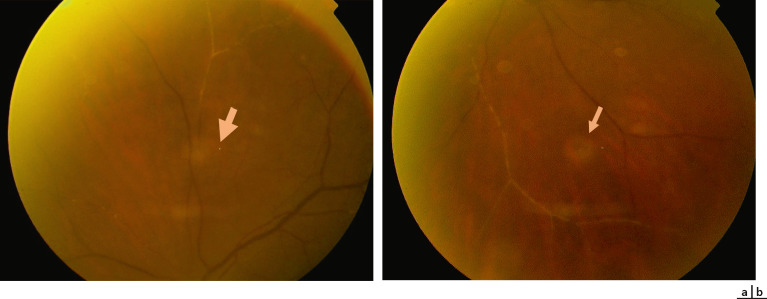
Fig. 16-26 a, b. Vasculopathie occlusive avec engainements vasculaires périphériques, responsables de lignes blanches continues (flèche).
Les séquelles après évolution de la maladie incluent cataracte, rubéose irienne et glaucome néovasculaire secondaire.
Le diagnostic, la prise en charge thérapeutique et le suivi seront aidés par la réalisation d'une angiographie à la fluorescéine. Elle permet de repérer les zones de non-perfusion, les néovaisseaux rétiniens ou papillaires (fig. 16-27).
- –Les néovaisseaux sont classiquement décrits en sea fan avec une hyperfluorescence intense aux temps artérioveineux et une exsudation aux temps tardifs. Les images en périphérie rétinienne montrent l'étendue des zones ischémiques, la présence d'engainements vasculaires et la présence éventuelle d'anévrismes, ainsi que la confirmation d'une occlusion de branche veineuse rétinienne.
- –Une angiographie de contrôle pourra déterminer l'efficacité de la photocoagulation au laser.
- –Une classification fondée sur les caractères cliniques a été proposée en 2004 par Saxena [15] :
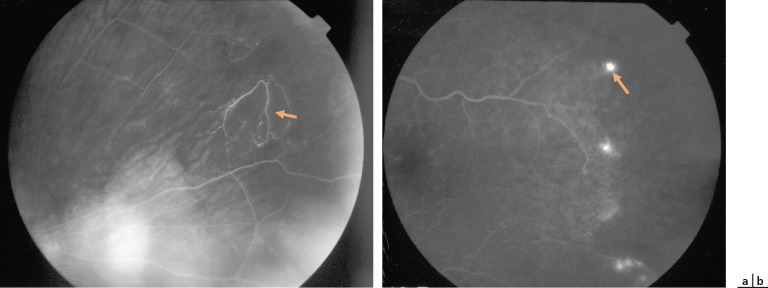
Fig. 16-27 a, b. Vasculopathie occlusive avec engainements vasculaires périphériques, responsables de lignes blanches continues (flèche).
Certains diagnostics différentiels et maladies associées doivent être éliminés lors de la présence d'une vascularite; maladie de Behçet, sclérose en plaques, lupus érythémateux disséminé, toxocarose, toxoplasmose, granulomatose de Wegener, lymphome, syphilis, borréliose de Lyme, tuberculose, voire sarcoïdose. La présence d'une hémorragie intravitréenne révélatrice doit faire éliminer d'autres causes : diabète, HTA, sarcoïdose, drépanocytose (voire autre hémoglobinopathie) ou une leucémie.
L'histoire naturelle de la maladie est marquée par une bilatéralisation. La complication la plus fréquente est l'hémorragie intravitréenne (49,32 % ) [16]. L'évolution de la maladie de Eales est assez variable, avec une rémission temporaire ou permanente dans certains cas et une progression continue pour d'autres.
La prise en charge thérapeutique est avant tout symptomatique. Le but est de : réduire l'inflammation vitréorétinienne; réduire le risque d'hémorragie intravitréenne provenant des néovaisseaux; prendre en charge une hémorragie intravitréenne et/ou des membranes épirétiniennes.
Les corticostéroïdes sont indiqués par voie orale ou péri-orbitaire aux stades de vascularite active avec périphlébites rétiniennes et hypersensibilité à la tuberculine. Une corticothérapie orale avec dose initiale élevée et décroissance progressive peut être instaurée dans ces cas. Les injections sous-ténoniennes sont également efficaces. Certains auteurs recommandent également des IVT de triamcinolone [17, 18]. Le bénéfice d'un traitement par Ozurdex (dispositif intravitréen de dexaméthasone) n'a pas encore été étudié dans cette indication [19].
En présence d'une hémorragie intravitréenne, il n'y a pas de recommandations formelles mais trois options sont possibles : l'observation, la chirurgie vitréorétinienne par vitrectomie ou les thérapies anti-VEGF. Les indications de la chirurgie par vitrectomie sont les suivantes : une hémorragie intravitréenne non résolutive spontanément (généralement après un délai de 3 mois) avec ou sans membrane épirétinienne, un DR tractionnel intéressant le pôle postérieur et un DR mixte et/ou rhegmatogène. L'apparition d'une cataracte est une complication reconnue de ce type de chirurgie [4].
La photocoagulation n'est pas recommandée au stade de vascularite. L'indication de la photocoagulation par laser concerne la maladie au stade proliférant avec néovaisseaux papillaires ou rétiniens actifs. Les zones de non-perfusion capillaire nécessitent un traitement par laser focal. Après photocoagulation, une régression de la néovascularisation rétinienne est observée dans 80 à 89 % des cas [20].
Il existe une association entre la maladie de Eales et la présence de VEGF [21]. Certains auteurs ont donc étudié le bénéfice d'un traitement par IVT de bévacizumab dans la régression des néovaisseaux et la réduction des hémorragies intravitréennes, mais les avis sont partagés.
Dans l'étude de Patwardhan [22], tous les patients inclus ayant bénéficié d'injections répétées de bévacizumab ont dÛ recevoir une vitrectomie. Ils ont également constaté la survenue de DR tractionnels. Aucun bénéfice postopératoire n'a été mis en évidence.
Kumar a publié des cas de DR rhegmatogènes chez des patients ayant bénéficié d'injections de bévacizumab en prétraitement dans des cas d'hémorragies intravitréennes ou de DR tractionnels [23].
Une prise en charge thérapeutique a été proposée sur la base de la classification établie en 2004 [15, 16] :
- –les stades 1a et 1b sont traités médicalement;
- –les stades 2a sont surveillés;
- –les stades 2b et 3a reçoivent une photocoagulation par laser argon;
- –les stades 3b sont surveillés en attendant que l'hémorragie du vitré se résolve;
- –les stades 4a sont opérés.
La maladie de Eales reste rare en pédiatrie. Elle est fréquemment révélée par une hémorragie intravitréenne spontanée. Il conviendra d'éliminer une tuberculose associée par la réalisation d'examens complémentaires tels qu'une intradermoréaction à la tuberculine et un quantiféron. Sa prise en charge pédiatrique impose une collaboration multidisciplinaire.
[1] Eales H Retinal haemorrhages associated with epistaxis and constipation Brim Med Rev: ( 1980 ) :9: 262-
[2] Wadsworth OF Recurrent retinal haemorrhage followed by the development of blood vessels in the vitreous Ophthalmol Rev: ( 1887 ) :6: 289-299
[3] Puttamma ST Varied fundus picture of central retinal vasculitis Trans Asia Pacific Acad Ophthalmol: ( 1970 ) :3: 520-
[4] Das T, Pathengay A, Hussain N, Biswas J Eales'disease : diagnosis and management Eye: ( 2010 ) :24: 472-482
[5] Rishi P, Rishi E, Gupta A Vitreous hemorrhage in children and adolescents in India J AAPOS: ( 2013 ) :17: 64-69
[6] Spirn MJ, Lynn MJ, Hubbard GB Vitreous hemorrhage in children Ophthalmology: ( 2006 ) :113: 848-852
[7] Saxena S, Srivastava P, Kumar D Decreased platelet membrane fluidity in retinal periphlebitis in Eales'disease Ocul Immunol Inflamm: ( 2006 ) :14: 113-116
[8] Sen A, Paine SK, Chowdhury IH Association of interferon-gamma, interleukin-10, and tumor necrosis factor-alpha gene polymorphisms with occurrence and severity of Eales'disease Invest Ophthalmol Vis Sci: ( 2011 ) :52: 171-178
[9] Singh R, Toor P, Parchand S Quantitative polymerase chain reaction for Mycobacterium tuberculosis in so-called Eales'disease Ocul Immunol Inflamm: ( 2012 ) :20: 153-157
[10] Rajpal , Singh UB, Mohapatra S Association of Mycobacterium tuberculosis in the causation of Eales'disease : an institutional experience Indian J Med Microbiol: ( 2015 ) :33: 43-45
[11] Das T, Biswas J, Kumar A Eales'disease Indian J Ophthalmol: ( 1994 ) :42: 3-18
[12] Atmaca LS, Idli A, Gunduz K Visualisation of retinal vasculitis in Eales disease Ocul Immunol Inflamm: ( 1993 ) :1: 41-48
[13] Atmaca LS, Batioglu F, Sonmez PA A long-term follow-up of Eales'disease Ocul Immunol Inflamm: ( 2002 ) :10: 213-221
[14] Abu El-Asrar AM, Al-Kharash SAI Full panretinal photocoagulation and early vitrectomy improve prognosis of retinal vasculitis associated with tuberculoprotein hypersensitivity (Eales'disease) Br J Ophthalmol: ( 2002 ) :86: 1248-1251
[15] Saxena S, Kumar D A new staging system for idiopathic retinal periphlebitis Eur J Ophthalmol: ( 2004 ) :14: 236-239
[16] Saxena S, Kumar D New classification system-based visual outcome in Eales'disease Indian J Ophthalmol: ( 2007 ) :55: 267-269
[17] Pathengay A, Pilli S, Das T Intravitreal triamcinolone acetonide in Eales'disease : a case report Eye: ( 2005 ) :19: 711-713
[18] Ishaq M, Feroze AH, Shahid M Intravitreal steroids may facilitate treatment of Eales'disease (idiopathic retinal vasculitis) : an interventional case series Eye: ( 2007 ) :21: 1403-1405
[19] Errera MH, Pratas A, Goldschmidt P Eales'disease J Fr Ophtalmol: ( 2016 ) :39: 474-482
[20] Gopal L, Abraham C Efficacy of photocoagulation in Eales'disease Trans Asia-Pacific Acad: ( 1985 ) :10: 689-
[21] Murugeswari P, Shukla D, Kim R Angiogenic potential of vitreous from proliferative diabetic retinopathy and Eales'disease patients PLoS One: ( 2014 ) :9: e107551-
[22] Patwardhan SD, Azad R, Shah BM, Sharma Y Role of intravitreal bevacizumab in Eales disease with dense vitreous hemorrhage: a prospective randomized control study Retina: ( 2011 ) :31: 866-870
[23] Kumar A, Sehra SV, Thirumalesh MB, Gogia V Secondary rhegmatogenous retinal detachment following intravitreal bevacizumab in patients with vitreous hemorrhage or tractional retinal detachment secondary to Eales'disease Graefes Arch Clin Exp Ophthalmol: ( 2012 ) :250: 685-690
S. Milazzo, N. Rahmania
L'incontinentia pigmenti (IP) est une maladie à hérédité dominante liée à l'X, dont la prévalence est de 1/500 000 et l'incidence de 0,0025 % [1, 2] chez les nouveau-nés. L'IP est aussi appelée Bloch-Sulzberger syndrome.
Cette pathologie atteint la peau, le système nerveux central, le squelette et les yeux [3, 4].
La mutation du gène IKBKG/NEMO situé sur le chromosome X, locus Xq28, est responsable de l'IP. Cette anomalie entraîne une perte de fonction du facteur nucléaire kappa-B (nuclear factor kappa-B ou NF-κB), responsable de l'augmentation de l'apoptose dans les tissus dérivant de l'ectoderme [5]. Quatre-vingt-dix-sept pour cent des cas sont des filles, car les mâles homozygotes pour la mutation meurent in utero, sauf les syndromes de Klinefelter qui peuvent développer une IP [6]. Les atteintes du système nerveux central ainsi que les lésions cutanées dépendent de la lyonisation du chromosome X [7]. Il existe des formes familiales et sporadiques [1].
Les lésions cutanées suivent les lignes de Blaschko et apparaissent vers l'âge de 6 mois dans 96 % des cas [3].
Quatre phases lésionnelles caractérisent l'IP et constituent les critères majeurs de la maladie :
- –stade 1 : il correspond à la phase inflammatoire et se caractérise par le développement de papules, de vésicules ou de pustules sur une base érythémateuse, distribuées de manière linéaire selon les lignes de Blaschko. Ces lésions peuvent être confondues avec un herpès ou un impétigo [2, 5, 7]. Les lésions sont principalement retrouvées au niveau des extrémités, mais peuvent aussi s'observer au niveau du cou et de la tête [8]. Chez plus de 90 % des patients, l'atteinte est présente à la naissance ou se développe durant les 2 premières semaines de vie puis disparaît vers l'âge de 4 mois [9];
- –stade 2 : il est aussi connu sous le nom de « phase verruqueuse » . Les lésions se distribuent également selon les lignes de Blaschko mais sont plus volontiers observées au niveau de la paume des mains et de la plante des pieds. Dans la plupart des cas, elles se développent entre 2 et 6 semaines de vie et disparaissent avant 6 mois [9];
- –stade 3 ou phase d'hyperpigmentation : les lésions sont brunâtres, linéaires et se distribuent au niveau du tronc, des extrémités, des plis cutanés de la tête et du cou (fig. 16-28). L'hyperpigmentation des mamelons, de l'aine et des aisselles est fréquente. Cette phase se développe durant les premiers mois de vie, disparaît à l'adolescence mais peut persister jusqu'à l'âge adulte, en général au niveau des aisselles et de l'aine. À ce stade, la biopsie révèle l'incontinence pigmentaire cutanée;
- –stade 4 : il est caractérisé par des plages d'hypopigmentation et d'atrophie et d'une absence de pilosité le plus souvent située au niveau des membres inférieurs. Ces lésions se développent pendant l'adolescence et peuvent devenir permanentes.
Il est rare d'observer chez un même patient la succession de tous les stades lésionnels [10].
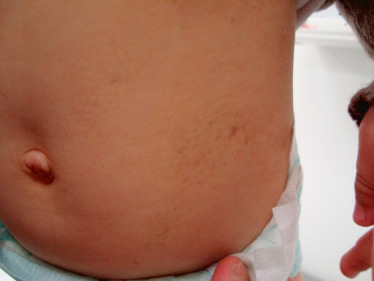
Fig. 16-28 Taches pigmentées, stade 3, situées sur l’abdomen.
Parmi les patients atteints de critères majeurs cutanés, 36,5 % des cas présentent une pathologie oculaire détectable dont 60 à 90 % de lésions rétiniennes [4].
L'atteinte ophtalmologique peut menacer le pronostic visual chez 20 % des patients atteints [7].
Les lésions rétiniennes, qui font toute la gravité de l'atteinte, sont secondaires à l'ischémie rétinienne périphérique et maculaire; ses conséquences sont la prolifération de néovaisseaux (avec ou sans saignement), une exsudation (fig. 16-29), une fibrose prérétinienne et un DR tractionnel [11]. À l'instar des lésions retrouvées dans la rétinopathie du prématuré, la persistance de la vascularisation rétinienne foetale est fréquente [2].
L'atteinte ophtalmologique la plus typique se caractérise par une masse rétrolentale secondaire à un DR. Elle est décrite dans 11,5 % des cas sous l'appellation tantôt de pseudo-gliome, tantôt de dysplasie rétrolentale [9].
D'autres anomalies au fond d'oeil telles que l'hypoplasie fovéolaire [12], le colobome ou l'atrophie optique sont également observables. Au sein d'une même famille dont les membres sont atteints d'IP, les manifestations rétiniennes de la maladie sont variables du fait de modifications épigénétique ou génique.
Chez un même patient, du fait du phénomène d'inactivation aléatoire du chromosome X à l'origine d'une expression phénotypique variable, l'atteinte est asymétrique entre les deux yeux [7].
En cas d'atteinte rétinienne, plusieurs études ont montré l'intérêt de la photocoagulation des zones ischémiques [13],
Le pronostic visuel est souvent bon chez les enfants ne présentant pas d'atteinte rétinienne au cours de la première année de vie [9],
Néanmoins, en cas d'atteinte rétinienne, la lésion peut rapidement évoluer vers le DR et la cécité, Toute suspicion diagnostique chez l'enfant doit conduire à la réalisation d'une angiofluorographie sous anesthésie générale dans les premières semaines de vie, La fréquence du suivi est ensuite déterminée par la sévérité de la rétinopathie avec espacement des contrôles si la maladie reste stable à partir de l'âge de 2 ans [7],
Au stade de DR, le traitement chirurgical est souvent peu efficace. Des atteintes non rétiniennes peuvent s'observer, comme la cataracte, le strabisme, le nystagmus, le ptosis, une microphtalmie, une pigmentation de la conjonctive, une altération cornéenne ou une myopie. Leur apparition est souvent plus tardive que les atteintes de la rétine [9].
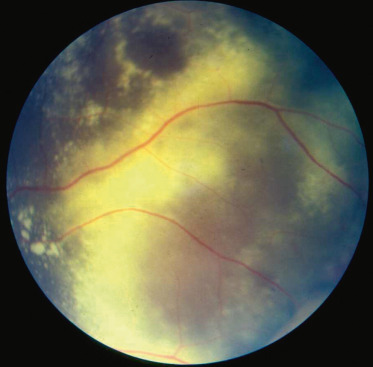
Fig. 16-29 Exsudation vasculaire majeure chez une enfant de 4 ans.
- –Neurologiques : comitialité, atteinte motrice, retard mental, microcéphalie. Les manifestations sont précoces. L'imagerie permet de mettre en évidence des plages de nécrose cérébrale, une atrophie, des lésions du corps calleux et d'autres lésions non spécifiques [14].
- –Dentaires : retard d'apparition, conisation (fig. 16-30), dystrophies, inclusions, oligodontie, déformation des incisives, fente palatine. La diminution d'excrétion salivaire doit être évaluée [15].
- –Phanères : alopécie, hypertrichose, dystrophie unguéale, syndactylie [1],
- –Squelettiques [1, 3, 4],
Fig. 16-30 Anomalies dentaires avec conisation.
Les critères diagnostiques sont résumés dans le Tableau 16-6.
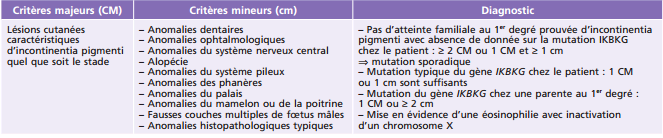
Il consiste à intervenir précocement par la mise en place d'examens :
- –ophtalmologiques pour dépister un DR;
- –neurologiques et neuropsychologiques à la recherche de crise d'épilepsie, de retard de développement avec bilan par IRM;
- –dentaires à partir de l'âge de 2 ans [3, 16].
Les lésions cutanées ne nécessitent aucun traitement.
Le diagnostic précoce de cette affection est essentiel. La découverte de cette maladie impose des examens successifs ophtalmologiques, neurologiques, dentaires et des tests génétiques, afin de prévenir des complications. La pertinence de ces éléments est confirmée dans le guide du National Institute of health (NIH), qui suggère un examen mensuel ophtalmologique avant l'âge de 4 mois [17]. Cette pathologie nécessite une prise en charge multidisciplinaire (dermatologue, neurologue, ophtalmologiste, etc.). Le diagnostic erroné de dermatite atopique peut conduire à la prescription d'une corticothérapie inutile et iatrogène chez le tout petit. Au total, c'est un diagnostic précoce et une surveillance étroite qui donnera à ces patients un meilleur pronostic.
[1] Fusco F, Paciolla M, Conte MI Incontinentia pigmenti : report on data from 2000 to 2013 Orphanet J Rare Dis: ( 2014 ) :9: 93-
[2] Poliak N, Le A, Rainey A Reticulated, hyperchromic rash in a striated pattern mimicking atopic dermatitis and fungal infection in a 2-month-old female: a case of incontinentia pigmenti Case Rep Pediatr: ( 2016 ) :2016: 9512627-
[3] Paller A, Mancini A, Hurwitz S :-
[4] Swinney CC, Han DP, Karth PA Incontinentia Pigmenti : a comprehensive review and update ophthalmic surg lasers imaging Retina: ( 2015 ) :46: 650-657
[5] Minic S, Trpinac D, Obradovic M Incontinentia pigmenti diagnostic criteria update Clinical Genetics: ( 2014 ) :85: 536-542
[6] Afshar H, Daneshpazhooh M, Kiani A Abnormal dentition in a boy with incontinentia pigmenti : case report Journal of Dentistry: ( 2012 ) :9: 267-270
[7] Chen CJ, Han IC, Goldberg MF Variable expression of retinopathy in a pedigree of patients with incontinentia pigmenti Retina: ( 2015 ) :35: 2627-2632
[8] Minic S, Obradovic M, Kovacevic I, Trpinac D Ocular anomalies in incontinentia pigmenti : literature review and meta-analysis Srp Arh Celok Lek: ( 2010 ) :138: 408-413
[9] Poziomczyk CS, Recuero JK, Bringhenti L Incontinentia pigmenti An Bras Dermatol: ( 2014 ) :89: 26-36
[10] Swamy DKN, Arunagirinathan AR, Krishnakumar AR, Sangili S Incontinentia pigmenti : a rare genodermatosis in a male child J Clin Diagn Res: ( 2015 ) :9: SD06-SD08
[11] Hadj-Rabia S, Froidevaux D, Bodak N Clinical study of 40 cases of incontinentia pigmenti Arch Dermatol: ( 2003 ) :139: 1163-1170
[12] Basilius J, Young MP, Michaelis TC Structural abnormalities of the inner macula in incontinentia pigmenti JAMA Ophthalmol: ( 2015 ) :133: 1067-1072
[13] Batioglu F, Ozmert E Early indirect laser photocoagulation to induse regression of retinal vascular abnormalities in incontinentia pigmenti Acta Ophthalmol: ( 2010 ) :88: 267-268
[14] Minic S, Trpinac D, Obradovic M Systematic review of central nervous system anomalies in incontinentia pigmenti Orphanet J Rare Dis: ( 2013 ) :8: 25-
[15] Hand JL What's new with common genetic disorders? Curr Opin Pediatr: ( 2015 ) :27: 460-465
[16] Incontinentia Pigmenti International Foundation IP Protocols :-
[17] Scheuerle AE, Ursini MV Incontinentia pigmenti Array:University of Washington ( 1999 ) - Seattle 1993-2016. Jun 8 [updated 2015 Feb 12]
M. Sampo, F. Matonti
Le syndrome de Goodpasture (syndrome pneumorénal) est également appelé glomérulonéphrite médiate antimembrane basale glomérulaire anti-glomerular basement membrane (anti-GBM) ou maladie à anticorps anti-GBM.
Cette affection auto-immune rare (0,5 à 1 cas/million d'habitants), associant une hémorragie pulmonaire à une glomérulonéphrite extracapillaire, est liée à la présence d'anticorps dirigés contre le domaine NC1 de la chaîne alpha-3 du collagène IV [1].
Ce syndrome atteint principalement les patients jeunes, avec une légère prédominance masculine [2].
II s'agit d'un syndrome pneumorénal.
L'atteinte pulmonaire se caractérise par un syndrome hémorragique alvéolaire, avec dyspnée, hémoptysies et anémie. Le scanner montre des opacités alvéolaires diffuses, et le lavage bronchio-alvéolaire confirme l'hémorragie alvéolaire. L'hémoptysie est le signe clinique le plus fréquent (82 à 98 % des cas) associé au syndrome de Goodpasture, tandis que les anomalies du fond d'œil ne concerneraient que 4 à 11 % des patients en fonction des séries [3, 4].
L'atteinte rénale se caractérise par une glomérulonéphrite extracapillaire rapidement progressive, conduisant à une insuffisance rénale aiguë puis chronique. La biopsie rénale permet la confirmation diagnostique, en retrouvant la présence de dépôts linéaires d'immunoglobulines G (IgG) le long des membranes basales glomérulaires en immunofluorescence. La présence d'anticorps anti-GBM circulants permet également de confirmer le diagnostic (présent dans 80 % des cas).
Au niveau oculaire, des anticorps anti-GBM circulants ont été retrouvés dans les vaisseaux choroïdiens, le corps ciliaire, la membrane de Bruch et au niveau de la capsule cristallinienne.
Le syndrome de Goodpasture peut être associé à des sclérites. Ainsi, Riono et al. ont rapporté un cas de sclérite nécrosante [5]. L'examen anatomopathologique retrouvait des zones d'inflammation granulomateuse nécrotique. Cet aspect de sclérite était similaire à celui observé avec d'autres pathologies systémiques, telles qu'une polyarthrite rhumatoïde, une polychondrite atrophiante, une maladie de Wegener.
Un cas d'uvéite associé à ce syndrome a été décrit. Il s'agissait d'un patient présentant une panuvéite granulomateuse avec présence d'infiltrats choroïdiens [6].
L'atteinte rétinienne au cours du syndrome de Goodpasture est rare. Ainsi, la présence d'hémorragies rétiniennes et d'exsudats a été retrouvée dans 4 à 11 % des patients selon les études [3, 4].
Un cas de néovascularisation choroï'dienne juxtapapillaire a été rapporté par Rowe et al. [7].
Des cas de DR exsudatifs ont été décrits [8-10]. L'apparition de ces DR serait liée aux dépôts d'IgG antimembrane basale au niveau de la membrane basale de l'épithélium pigmentaire et des vaisseaux choroïdiens, ce qui entraînerait une rupture de la BHR externe et l'apparition d'une exsudation. L'apparition de ces DR exsudatifs pourrait être également favorisée par un contexte hypertensif.
L'origine de l'atteinte ophtalmologique rétinienne (hémorragies, exsudats, DR exsudatif) dans le cadre du syndrome de Goodpasture demeure discutée et pourrait être en rapport avec l'HTA ou la positivité des anti-neutrophil cytoplasmic antibodies (ANCA), plutôt qu'une lésion directe par les anticorps anti-GBM [6].
L'atteinte rétinienne survient habituellement après l'apparition des signes pulmonaires et rénaux, mais de façon exceptionnelle, l'atteinte ophtalmologique peut les précéder. Ainsi, Köhler rapportait un cas ayant présenté une vascularite rétinienne associé à un œdème maculaire, avant l'apparition de l'hémoptysie et d'une insuffisance rénale [11].
Le syndrome de Goodpasture est une urgence médicale, puisqu'en l'absence de traitement, cette pathologie peut être fatale. Le traitement systémique repose sur des corticoïdes, immunosuppresseurs, échanges plasmatiques. La précocité du traitement est un élément pronostique important, permettant d'éviter l'insuffisance rénale et l'insuffisance respiratoire.
[1] Beauvillard D, Segalen I, Le Meur Y Auto-anticorps anti-membrane basale glomérulaire et syndrome de Goodpasture Immuno analyse Bio Spe: ( 2011 ) :26: 60-67
[2] Bolton WK Goodpasture's syndrome Kidney Int: ( 1996 ) :50: 1753-1766
[3] Proskey AJ, Weatherbee L, Easterling RE Goodpasture's syndrome. A report of five cases and review of the literature Am J Med: ( 1970 ) :48: 162-173
[4] Benoit FL, Rulon DB, Theil GB Goodpasture's syndrome : clinicopathologic entity Am J Med: ( 1964 ) :37: 424-444
[5] Riono WP, Hidayat AA, Rao NA Scleritis : a clinicopathologic study of 55 cases Ophthalmology: ( 1999 ) :106: 1328-1333
[6] Chak M, Stanford MR, Poon W Uveitis initiating an auto-immune reaction resulting in Goodpasture's syndrome in a Chinese man Br J Ophthalmol: ( 2002 ) :86: 1188-1190
[7] Rowe PA, Mansfield DC, Dutton GN Ophthalmic features of fourteen cases of Goodpasture's syndrome Nephron: ( 1994 ) :68: 52-56
[8] Jampol LM, Lahov M, Albert DM, Craft J Ocular clinical findings and basement membrane changes in Goodpasture's syndrome Am J Ophthalmol: ( 1975 ) :79: 452-463
[9] Boucher MC, el Toukhy EA, Cormier G Bilateral serous retinal detachments associated with Goodpasture's syndrome Can J Ophthalmol: ( 1998 ) :33: 46-49
[10] Hoscheit AM, Austin JK, Jones WL Nonrhegmatogenous retinal detachment in Goodpasture's syndrome : a case report and discussion of the clinicopathologic entity J Am Optom Assoc: ( 1993 ) :64: 563-567
[11] Köhler U Unusual course of Goodpasture syndrome Klin Monbl Augenheilkd: ( 1988 ) :192: 354-357
M. Chardavoine, F. Matonti
Selon la nomenclature de Chapel Hill, le syndrome de Churg et Strauss (SCS) est une vascularite des vaisseaux de petit calibre, soit artérioles et veinules, fréquemment associée à de l'asthme et une hyperéosinophilie.
Elle porte le nom de deux pathologistes, J. Churg et L. Strauss, qui ont été les premiers à la décrire [1-5].
Sa prévalence est d'environ 1 sur 100 000. La maladie touche aussi bien les hommes que les femmes et apparaît généralement chez les adultes (incidence maximale entre 30 et 50 ans) [5]. Elle survient exceptionnellement chez les enfants [6-12].
II s'agit d'une maladie auto-immune. Chez la plupart des patients, le SCS évolue en trois phases :
- –initialement, on diagnostique un asthme et différentes manifestations allergiques telles que les rhinites et les sinusites;
- –l'hyperéosinophilie sanguine apparaît ensuite, il en découle l'infiltration d'éosinophiles surtout au niveau des poumons, du cœur et du tractus gastro-intestinal;
- –plusieurs années après le début des symptômes, la vascularite nécrosante systémique survient [4, 5, 13, 14].
Les critères de classification du SCS de l'American College of Rheumatology (ACR) de 1990 sont : asthme, éosinophilie sanguine> 10 % , mono- ou polyneuropathie, infiltrats pulmonaires labiles, sinusite, présence d'éosinophiles extravasculaires à la biopsie. La présence de quatre de ces six critères permet le classement comme syndrome de Churg et Strauss avec une sensibilité de 85 % et une spécificité de 99,7 % [2].
Au bilan biologique, on observe dans 60 % des cas la présence d'anticorps anticytoplasme des polynucléaires neutrophiles (ANCA) ayant une fluorescence périnucléaire (perinuclear anti-neutrophil cytoplasmic antibodie [pANCA]) et pour cible la myélopéroxydase (MPO) [15-17]. Le facteur rhumatoïde est positif dans environ 50 % des cas [14].
On retrouve également une hyperéosinophilie (généralement> 1500/mm) et un syndrome inflammatoire biologique (vitesse de sédimentation augmentée chez 80 % des patients) [12].
Les constatations anatomopathologiques montrent trois types de lésions élémentaires dans le SCS : vascularite nécrosante touchant les artères et les veines de petit calibre; infiltration éosinophilique tissulaire; granulomes extravasculaires contenant une nécrose centrale entourée de cellules épithélioïdes.
Le SCS est une maladie avec atteinte multiviscérale [1, 18, 19].
L'atteinte pulmonaire est constante, Nous relatons principalement de l'asthme, mais aussi une hémorragie alvéolaire, une pleurésie, des infiltrats pulmonaires,
Les manifestations ORL sont habituelles, mais de bon pronostic : sinusite, rhinite allergique, polypose sinusienne [12, 17].
L'atteinte neurologique est très fréquente (55 à 75 % ). Elle peut être périphérique, sous la forme d'une neuropathie, ou centrale, liée à la vascularite cérébrale [4, 12],
L'atteinte cutanée est présente dans 40 à 70 % des cas [4, 12, 17, 20], Elle se résume en un purpura vasculaire, des papules et nodules, des lésions urticariennes, un livedo reticularis, un syndrome de Raynaud [12, 20],
Chez 20 % des malades, on retrouve une atteinte cardiaque, principalement myocardite et péricardite. Elle représente la première cause de mortalité [1, 16, 17, 19, 21].
Le SCS peut induire une atteinte digestive, de mauvais pronostic, dans 30 % des cas : douleurs abdominales, méléna, perforation digestive [12, 22],
L'atteinte rénale est rare, à type de glomérulonéphrite extracapillaire, Elle est associée à la présence d'ANCA anti-MPO [4, 12],
On observe également des arthralgies et des myalgies [12].
L'atteinte ophtalmologique est possible mais rare au cours du SCS, Un cas chez un enfant de 10 ans a été décrit [9], Les atteintes suivantes sont rapportées : nodules palpébraux ou conjonctivaux à type de granulomes à éosinophiles, épisclérites, sclérites antérieures, ulcérations cornéennes, uvéites, vascularites rétiniennes, choroïdite, occlusion de l'artère centrale de la rétine, occlusion de branche artérielle, neuropathie optique ischémique antérieure aiguë, neuropathie optique [4, 5, 9, 23–27]. Une atteinte orbitaire sous la forme d'une pseudo-tumeur inflammatoire de l'orbite a également été décrite [27, 28],
Le five factors score (FFS) est un score pronostique qui peut s'appliquer à la panartérite noueuse, la micropolyangéite, le SCS et la maladie de Wegener, Il est composé :
- –de quatre facteurs « péjoratifs » (+ 1 point) : âge> 65 ans; atteinte cardiaque; atteinte digestive; atteinte rénale;
- –d'un facteur « protecteur » (– 1 point) : atteinte ORL,
Le pronostic du SCS a été transformé depuis l'apparition des corticoïdes, La corticothérapie est la base de la thérapeutique, parfois associée aux immunosuppresseurs [12, 13],
[1] Churg J, Strauss L Allergic granulomatosis, allergic angiitis, and periarteritis nodosa Am J Pathol: ( 1951 ) :27: 277-301
[2] Masi A, Hunder G, Lie J The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis angiitis) Arthritis Rheum: ( 1990 ) :33: 1094-1100
[3] Taniguchi M, Tsurikisawa N, Higashi N Treatment for Churg-Strauss syndrome : induction of remission and efficacy of intravenous immunoglobulin therapy Allergol Int: ( 2007 ) :56: 97-103
[4] Safran AB, Vighetto A, Landis T, Cabanis E Pathologie inflammatoire Neuroophtalmologie. Rappord de la Société française d'ophtalmologie: ( 2004 ) 579-581 Chap. 5
[5] Pournaras CJ Mécanismes occlusifs vasculaires. Pathologie pariétale Pathologies vasculaires oculaires. Rappord de la Société française d'ophtalmologie: ( 2008 ) 152-153 Chap. 6
[6] Gendelman S, Zeft A, Spalding SJ Childhood-onset eosinophilic granulomatosis with polyangiitis (formerly Churg-Strauss syndrome) : a contemporary single-center cohort J Rheumatol: ( 2013 ) :40: 929-935
[7] Boyer D, Vargas SO, Slattery D Churg-Strauss syndrome in children: a clinical and pathologic review Pediatrics: ( 2006 ) :118: e914-e920
[8] Wang SJ, Yang YH, Lin YT Childhood Churg-Strauss syndrome : report of a case J Microbiol Immunol Infect: ( 2000 ) :33: 263-266
[9] Dillon MJ Rare vasculitic syndromes Ann Med: ( 1997 ) :29: 175-179
[10] Partal A, Moshfeghi DM, Alcorn D Churg-Strauss syndrome in a child : retina and optic nerve findings Br J Ophthalmol: ( 2004 ) :88: 971-972
[11] El-Gamal Y Churg-strauss syndrome in the pediatric age group World Allergy Organ J: ( 2008 ) :1: 34-40
[12] Guillevin L, Cohen P, Gayraud M Churg-Strauss syndrome. Clinical study and long-term follow-up of 96 patients Medicine (Baltimore): ( 1999 ) :78: 26-37
[13] Guillevin L, Lhote F, Gherardi R Polyarteritis nodosa, microscopic polyangiitis, and Churg-Strauss syndrome : clinical aspects, neurologic manifestations, and treatment Neurol Clin: ( 1997 ) :15: 865-886
[14] Pagnoux C, Guilpain P, Guillevin L Churg-Strauss syndrome Curr Opin Rheumatol: ( 2007 ) :19: 25-32
[15] Kallenberg CG Antineutrophil cytoplasmic antibodies (ANCA) and vasculitis Clin Rheumatol: ( 1990 ) :9: 132-135
[16] Haas C, Géneau C, Odinot J L'angéïte allergique avec granulome : syndrome de Churg et Strauss. Étude rétrospective de 16 observations Ann Med Interne (Paris): ( 1991 ) :142: 335-342
[17] Leatherman J, Davies S, Hoidal J Alveolar hemorrhage syndromes : diffuse microvascular lung hemorrhage in immune and idiopathic disorders Medicine: ( 1984 ) :63: 343-361
[18] Guillevin L, Guittard T, Bletry O Systemic necrotizing angiitis with asthma : causes and precipitating factors in 43 cases Lung: ( 1987 ) :165: 165-172
[19] Comarmond C, Pagnoux C, Khellaf M, French Vasculitis Study Group Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) : clinical characteristics and long-term followup of the 383 patients enrolled in the French Vasculitis Study Group cohort Arthritis Rheum: ( 2013 ) :65: 270-281
[20] Davis MD, Daoud MS, McEvoy MT, Su WP Cutaneous manifestations of Churg-Strauss syndrome: a clinicopathologic correlation J Am Acad Dermatol: ( 1997 ) :37: 199-203
[21] Le Thi Huong Du, Guillevin L, Godeau P Prognostic factors in periarteritis nodosa and Churg-Strauss angiitis. Multifactor study of the survival of 165 patients Presse Med: ( 1985 ) :14: 1341-
[22] Guillevin L, Lhote F, Gallais V Gastrointestinal tract involvement in polyarteritis nodosa and Churg-Strauss syndrome Ann Med Interne (Paris): ( 1995 ) :146: 260-267
[23] Nissim F, Von der Valde J, Czernobilsky B A limited form of Churg-Strauss syndrome : ocular and cutaneous manifestations Arch Pathol Lab Med: ( 1982 ) :106: 305-307
[24] Vitali C, Genovesi-Ebert F, Romani A Ophthalmological and neuro-ophthalmological involvement in Churg-Strauss syndrome : a case report Graefes Arch Clin Exp Ophthalmol: ( 1996 ) :234: 404-408
[25] Shields CL, Shields JA, Rozanski TI Conjunctival involvement in Churg-Strauss syndrome Am J Ophthalmol: ( 1986 ) :102: 601-605
[26] Robin JB, Schanzlin DJ, Meisler DM Ocular involvement in the respiratory vasculitides Surv Ophthalmol: ( 1985 ) :30: 127-140
[27] Takanashi T, Uchida S, Arita M Orbital inflammatory pseudotumor and ischemic vasculitis in Churg-Strauss syndrome: report of two cases and review of the literature Ophtalmology: ( 2001 ) :108: 1129-1133
[28] McNab AA Orbital inflammation in Churg-Strauss syndrome Orbit: ( 1998 ) :17: 203-205
P. Gascon, F. Matonti
Mikito Takayasu a décrit en 1908, pour la première fois, cette artérite (dite artérite de Takayasu ou TAK) chez une jeune femme se plaignant de baisse d'acuité visuelle (BAV) bilatérale. Il avait observé une anastomose artérioveineuse en couronne autour du nerf optique qui représente en réalité un stade avancé de la maladie [1]. Également appelée « maladie des femmes sans pouls » , il s'agit d'une inflammation granulomateuse chronique de l'aorte et de ses branches principales provoquant un rétrécissement sténotique puis une occlusion vasculaire.
II s'agit d'une pathologie à prépondérance féminine (sex-ratio 1:9) touchant des patientes jeunes (autour de 20 ans) [2]. L'incidence varie en fonction de la localisation géographique mais reste très faible : elle est par an de 2,6/million d'habitants en Amérique du Nord, 1,2/million au Japon et inférieure à 1/million en Europe.
L'association génétique la plus retrouvée est avec l'antigène des leucocytes humains (human leucocyte antigen [HLA]) B52:01.
Lors de la phase pré-occlusive, on retrouve des signes systémiques non spécifiques. Le diagnostic est porté en moyenne 10 mois après le début de l'atteinte inflammatoire, lors de la phase occlusive ischémique de l'aorte et de ses branches principales.
Bien qu'une analyse histologique soit préférable, l'ACR a établi des critères diagnostiques [3]. La présence d'au moins trois des six critères suivants permet de classer une vascularite en maladie de Takayasu (sensibilité = 90,5 % et spécificité = 97,8 % ) :
- –âge de survenue inférieur à 40 ans;
- –claudication vasculaire des extrémités;
- –diminution d'au moins 1 pouls brachial;
- –différence de pression artérielle systolique de plus de 10 mmHg entre les deux bras;
- –souffle sur l'artère sous-clavière ou l'aorte abdominale;
- –anomalies artériographiques : sténose ou occlusion de l'aorte ou de ses branches.
Les manifestations oculaires sont décrites dans 8,1 à 68 % des cas. La principale atteinte est rétinienne (jusqu'à 35 % des cas) et l'amaurose fugace est le symptôme fonctionnel le plus fréquent [4], à côté des douleurs oculaires ou encore de la BAV progressive.
Les atteintes rétiniennes sont principalement représentées par la rétinopathie hypertensive (RH) suivie de la rétinopathie de Takayasu (RT). Pour l'ensemble des patients examinés par Chun et al. [4], il y avait environ 1 patient sur 3 atteints de RH et 13,5 % des patients avaient une RT. Lorsque les artères rénales sont touchées, une HTA rénovasculaire est responsable de RH; alors que pour le développement de la RT, il s'agit d'une sténose carotidienne provoquant une hypoperfusion chronique oculaire. Cette dernière est dépendante du nombre, du degré et de la durée des sténoses des artères cervicales, ainsi que du développement ou non d'un réseau vasculaire collatéral de suppléance.
Uyama et al. [1] ont classé la RT en quatre stades :
- –stade 1 : dilatations veineuses rétiniennes;
- –stade 2 : micro-anévrismes rétiniens initialement au pôle postérieur puis en périphérie;
- –stade 3 : anastomoses artérioveineuses en moyenne périphérie ou en péripapillaire;
- –stade 4 : complications oculaires secondaires à l'ischémie chronique étendues (néovascularisation rétinienne, hémorragie intravitrénne, rubéose irienne, glaucome néovasculaire).
Il existe des manifestations plus rares comme des cas de : occlusions de branches artérielles rétiniennes [5]; occlusions de branches veineuses rétiniennes; neuropathie optique ischémique antérieure (NOIA); sclérites; uvéites [6-8]; neuropathie optique glaucomateuse. Une cataracte, en plus de celle sous-capsulaire postérieure cortico-induite, peut être retrouvée chez les patients atteints de TAK.
Les premiers clichés angiographiques à la fluorescéine (AF) d'une atteinte rétinienne de la maladie de Takayasu ont été réalisés en 1965. Des micro-anévrismes et des zones rétiniennes de non-perfusion ont été retrouvés. Sur les temps très précoces de l'AF, Tanaka et al. ont mis en évidence un retard du colorant dans les lobules de la choriocapillaire, confirmé en angiographie au vert d'indocyanine par un ensemble de taches hypocyanescentes. L'AF permet également de mettre en évidence un allongement du temps bras-rétine, un retard de remplissage artérioveineux, des shunts artérioveineux et des néovaisseaux prérétiniens [4].
Bien qu'il s'agisse d'une maladie inflammatoire, la TAK n'est parfois pas associée à une élévation de la C-reactive proteine (CRP) et/ou de la vitesse de sédimentation. Différentes équipes ont alors trouvé des marqueurs d'activité de la maladie, plus fiable, comme la matrix metalloproteinase 2, 3 et 9 (MMP-2, -3, -9), le dosage sérique de l'interleukine 6 (IL-6) ou encore la pentraxin 3 (PTX-3).
L'anatomopathologie montre au niveau artériel les séquelles d'une inflammation prolongée. On retrouve un infiltrat inflammatoire lymphocytaire ou gigantocellulaire de la média et de l'adventice autour des vasa vasorum. La média et l'intima sont épaissies par accumulation de mucopolysaccharides et infiltration de myofibroblastes. La paroi artérielle est le siège d'une fibrose et d'une destruction du tissu élastique entraînant sa sténose et sa thrombose.
Dans de rares cas, la TAK peut toucher des vaisseaux plus petits entraînant des vascularites nécrosantes cutanées, une myocardite ou des atteintes rétiniennes [5].
La TAK est une vascularite des gros vaisseaux comme la maladie de Horton. Il s'agirait de deux expressions distinctes d'un même processus physiopathologique.
Il faut également envisager toutes les autres causes d'aortites et de maladie de la paroi aortique, qu'elles soient infectieuses, inflammatoires ou développementales.
En phase inflammatoire, une corticothérapie systémique doit être installée en première intention, avec une dose oscillant entre 0,5 mg et 1 mg/kg/j. Elle permet dans un premier temps une rémission chez environ 60 % des patients. En cas de réponse clinique insuffisante ou d'apparition d'une rechute lors de la décroissance de la corticothérapie, on peut être amené à utiliser un deuxième immunosuppresseur (méthotrexate, azathioprine, ciclosporine, mycophénolate mophétil), permettant de mieux contrôler la pathologie [2].
Chez les patients qui restent réfractaires à la corticothérapie et aux immunosuppresseurs classiques, les anti-tumor necrosis factor α ou anti-TNF-α (étanercept et infliximab) semblent être une alternative thérapeutique prometteuse.
Le tocilizumab (anticorps monoclonal antirécepteur de l'IL-6) semble être une biothérapie intéressante dans le contrôle de la TAK, comme dans la maladie de Horton [9], avec en plus une bonne tolérance.
À côté de ces biothérapies, un traitement anti-hypertenseur est instauré en cas d'HTA.
Si l'on ne traite pas des sténoses radiologiques, qui peuvent être dans certaines situations chroniques sans retentissement d'aval grâce au développement de collatérales, il est indispensable de revasculariser toute sténose ayant un retentissement clinique significatif, un risque de rupture ou générant une insuffisance aortique sévère. Que ce soit pour l'angioplastie ou pour la chirurgie, ces gestes de revascularisation doivent être effectués à distance d'une poussée inflammatoire pour éviter un risque trop important de resténose.
Un traitement ophtalmologique n'est envisagé qu'en cas de complication secondaire à une hypoperfusion chronique (panphotocoagulation rétinienne, vitrectomie, cryo-cyclo-destruction rétinienne). En cas d'amaurose fugace associée à une NOIA dans un contexte de TAK, une corticothérapie peut rapidement être instaurée, afin d'éviter une cécité [5].
Le pronostic de la TAK s'est largement amélioré au cour de ces dernières années. Sur 108 patients, Park [10] a retrouvé une survie tous cas confondus de 92,9 % à 5 ans et de 87,2 % à 10 ans. Cependant le pronostic reste dépendant de la présence et du nombre de facteur de risque (insuffisance cardiaque, accident vasculaire cérébral, RT, HTA rénovasculaire). Les patients avec une RT ont ainsi une mortalité plus élevée que ceux qui n'en n'ont pas.
Au niveau ophtalmologique, l'acuité visuelle (AV) est relativement conservée sauf en cas de RT stade 4 (AV moyenne de 1/10), donc lors de complications secondaires à l'ischémie rétinienne chronique [4].
La TAK est classiquement une pathologie des gens de moins de 40 ans. Il n'est pas rare de voir des enfants ou des adolescents atteints de cette pathologie, puisqu'il s'agit de la troisième cause de vascularite en pédiatrie. Le cas le plus jeune est un enfant de 6 mois.
La TAK de l'enfant (TAK-e), comme celle de l'adulte, est principalement localisée au niveau de la crosse aortique chez les Japonais, alors qu'en Inde et chez les TAK-e d'Amérique du Nord, il s'agit surtout d'une atteinte de l'aorte thoracique et abdominale [11].
Chez les enfants aussi, il existe une plus grande majorité de cas féminins, même si le sex-ratio (environ 3:1) semble un peu moins prononcé que chez l'adulte [11, 12]. L'incidence annuelle exacte du TAK-e est difficile à estimer, cependant la plupart des études européennes et d'Amérique du Nord trouvent une incidence globale annuelle entre 1 et 2,6/1 000 000 habitants.
La classification pour la TAK-e, selon European League Against Rheumatis (EULAR)/Pediatric Rheumatology International Trials Organization (PRINTO)/Pediatric Rheumatology European Society (PRES), classe la pathologie avec une sensibilité de 100 % et une spécificité de 99,9 % (Tableau 16-7) [13].
Tableau 16-7 – Classification de l’artérite de Takayasu chez l’enfant selon EULAR/PRINTO/PRES.

CRP : C-reactive proteine ; EULAR : European League Against Rheumatis ; IRM : imagerie par résonance magnétique ; PRES : Pediatric Rheumatology European Society ; PRINTO : Pediatric Rheumatology International Trials Organization ; TA : tension artérielle ; TDM : tomodensitométrie ; VS : vitesse de sédimentation.
Lors de la phase inflammatoire pré-occlusive, les signes systémiques sont non spécifiques. On retrouve de la fièvre, une anorexie, des sueurs, des arthralgies, des éruptions cutanées, une perte de poids, etc. Cela entraîne fréquemment un retard diagnostic allant de 2 à 11 ans (4 fois plus élevée que chez l'adulte), qui peut être à l'origine de séquelles vasculaires jusque chez un tiers des enfants à un stade avancé de TAK-e.
Le principal symptôme général est l'HTA (de 57 à 93 % selon les études), principalement par atteinte rénovasculaire.
Les manifestations secondaires à l'ischémie par sténose vasculaire sont principalement représentées par l'absence de pouls périphériques. On pourra ensuite retrouver les souffles vasculaires et les claudications des membres mais de manière plus rare, à la différence de l'adulte.
L'atteinte oculaire est plus rare chez l'enfant [14]. Dans une série de 17 enfants atteints de TAK, de 5 à 11 ans, seuls 5 enfants ont eu une atteinte ophtalmologique, à type de RH [15].
Becker et al. [6] ont publié le cas d'une enfant de 5 ans dont la première manifestation du TAK-e était une uvéite antérieure aiguë hypertensive, compliquée par la suite d'un glaucome et d'une cataracte cortico-induite. Kausman [7] a également décrit un cas de panuvéite chez un enfant de 12 ans atteint de TAK.
Un autre cas, chez un enfant de 12 ans avec TAK, avec atteinte ophtalmologique antérieure et postérieure a été décrit [8]. Cet enfant s'est initialement présenté avec une uvéite antérieure bilatérale sans BAV. L'évolution a été marquée par l'apparition d'un œdème maculaire cystoïde bilatéral, associé cette fois à une BAV, objectivé par une angiographie à la fluorescéine sans mise en évidence de ralentissement circulatoire rétinien. Les manifestations oculaires ont par la suite bien répondu à un traitement anti-inflammatoire local et général, malgré quelques épisodes de récidives de l'atteinte antérieure.
Les marqueurs biologiques de l'inflammation sont les mêmes que chez l'adulte avec également un intérêt particulier pour la vitesse de sédimentation (VS) et la CRP qui rentre dans les critères diagnostiques « mineurs » du TAK-e. Comme nous l'avons vu dans le Tableau 16-7 , un examen d'imagerie vasculaire d'exploration des gros vaisseaux est nécessaire au diagnostic de TAK-e. Cet examen recherchera anévrismes, dilatations, obstructions ou sténoses artérielles.
Comme chez l'adulte, à la phase inflammatoire initiale les traitements de première intention sont les corticoïdes plus ou moins associés à des immunosuppresseurs de deuxième ligne, avec pour objectif de limiter les séquelles vasculaires sténosantes.
Dans une série de six cas de TAK-e, Ozen et al. [16] ont utilisé avec succès du cyclophosphamide en association avec des corticoïdes avec ensuite un relais par méthotrexate. En effet, une corticothérapie au long cours peut avoir chez l'enfant des conséquences graves notamment pour la croissance. L'intérêt des anti-TNF-a doit encore être évalué chez l'enfant mais la perspective de leurs utilisations et les premiers essais semblent intéressants.
Le pronostic global de la maladie a pu bénéficier des progrès sur la connaissance physiopathologique de la maladie. En 1991, une étude a retrouvé un taux de mortalité de 35 % à 5 ans sur 26 enfants atteints de TAK [17], alors qu'en 2014 Goel et al. [11] n'ont eu qu'un seul décès sur 40 TAK-e, de même que dans une autre étude récente brésilienne (2016), il n'y a eu que 5 décès sur les 71 enfants suivis [12].
[1] Uyama M, Asayama K Retinal vascular changes in Takayasu's disease (pulseless disease), occurrence and evolution of the lesion International Symposium on Fluorescein Angiography Ghent 28 March-1 April 1976. Volume 9 of the series Documenta Ophthalmologica Proceedings Series:Springer ( 1976 ) 549-554
[2] Sugiyama K, Ijiri S, Tagawa S, Shimizu K Takayasu disease on the centenary of its discovery Jpn J Ophthalmol: ( 2009 ) :53: 81-91
[3] Arend WP, Michel BA, Bloch DA The American College of Rheumatology 1990 criteria for the classification of Takayasu arteritis Arthritis Rheum: ( 1990 ) :33: 1129-1134
[4] Chun YS, Park SJ, Park IK The clinical and ocular manifestations of Takayasu arteritis Retina: ( 2001 ) :21: 132-140
[5] Noel N, Butel N, Le Hoang P Small vessel involvement in Takayasu's arteritis Autoimmun Rev: ( 2013 ) :12: 355-362
[6] Becker RW, Sohn RL, Poulik JM, Berguer R Takayasu's arteritis presenting as uveitis in a 5-year-old girl Ann Vasc Surg: ( 2005 ) :19: 258-262
[7] Kausman JY, Walker A, Piper S Acute panuveitis and Takayasu's arteritis Arch Dis Child: ( 2003 ) :88: 938-939
[8] McDonald MA, Ojaimi E, Favilla I Anterior uveitis in a child with Takayasu's arteritis Clin Experiment Ophthalmol: ( 2004 ) :32: 336-339
[9] Salvarani C, Magnani L, Catanoso M Tocilizumab : a novel therapy for patients with large-vessel vasculitis Rheumatol Oxf Engl: ( 2012 ) :51: 151-156
[10] Park MC, Lee SW, Park YB Clinical characteristics and outcomes of Takayasu's arteritis : analysis of 108 patients using standardized criteria for diagnosis, activity assessment, and angiographic classification Scand J Rheumatol: ( 2005 ) :34: 284-292
[11] Goel R, Kumar TS, Danda D Childhood-onset Takayasu arteritis – experience from a tertiary care center in South India J Rheumatol: ( 2014 ) :41: 1183-1189
[12] Clemente G, Hilario MO, Len C Brazilian multicenter study of 71 patients with juvenile-onset Takayasu's arteritis : clinical and angiographic features Rev Bras Reumatol: ( 2016 ) :56: 145-151
[13] Ozen S, Pistorio A, Iusan SM EULAR/PRINTO/PRES criteria for Henoch – Schonlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis : Ankara 2008. Part II : final classification criteria Ann Rheum Dis: ( 2010 ) :69: 798-806
[14] Peter J, David S, Danda D Ocular manifestations of Takayasu arteritis : a cross-sectional study Retina: ( 2011 ) :31: 1170-1178
[15] Muranjan MN, Bavdekar SB, More V Study of Takayasu's arteritis in children : clinical profile and management J Postgrad Med: ( 2000 ) :46: 3-8
[16] Ozen S, Duzova A, Bakkaloglu A Takayasu arteritis in children : preliminary experience with cyclophosphamide induction and corticosteroids followed by methotrexate J Pediatr: ( 2007 ) :150: 72-76
[17] Morales E, Pineda C, Martinez-Lavin M Takayasu's arteritis in children J Rheumatol: ( 1991 ) :18: 1081-1084
S. Milazzo, N. Taright
L'anémie est un motif fréquent de consultation en pédiatrie. Elle est définie par une diminution du taux d'hémoglobine (Hb) en dessous de deux déviations standard (– 2 DS) par rapport à la moyenne pour l'âge [1]. Il faut tenir compte dans l'interprétation du chiffre d'hémoglobine, de l'état d'hémodilution ou d'hémoconcentration de l'enfant. Les signes cliniques de l'anémie sont la conséquence de l'hypoxie tissulaire et des mécanismes d'adaptation. Le praticien doit rechercher cliniquement une asthénie, une dyspnée d'effort, une pâleur cutanéomuqueuse, un souffle systolique, une cassure de la courbe staturopondérale. La gravité de l'anémie est évaluée par la rapidité d'installation, la tolérance cardiovasculaire et l'état de conscience associé à un possible trouble du comportement de l'enfant.
L'orientation étiologique est guidée par la connaissance des trois mécanismes de l'anémie :
- –défaut de production de globules rouges, souvent carentielle;
- –destruction augmentée de globules rouges par hémolyse;
- –perte de globules rouges, par hémorragie.
Les étiologies sont donc :
- –les anémies microcytaires/hypochromes non régénératives : carence martiale, syndrome inflammatoire, intoxication au plomb (saturnisme), syndromes thalassémiques;
- –les anémies normo- ou macrocytaires, normochromes aré-génératives : anémie mégalobastique (carence en folates et vitamine B), syndrome de Pearson, érythroblastopénie, moelle éry-throblastique et dysérythropoïétique, moelle pauvre ou désertique (aplasie médullaire, myélofibrose aiguë), moelle envahie (hémopathies malignes, métastases de tumeurs solides, syndrome d'activation macrophagique), hypersplénisme, insuffisance rénale, endocrinopathies (hypothyroïdie, insuffisance surrénale, hypogonadisme, hypopituitarisme, hyperparathyroïdie), hémorragie aiguë récente;
- –les anémies normochromes normocytaires régénératives :
Les signes suivants peuvent s'associer à l'anémie :
- –des signes résultant de la chute du taux d'hémoglobine et ses répercussions organiques;
- –des signes spécifiques à un type particulier d'anémie telle la drépanocytose;
- –des signes d'accompagnement en lien avec une pathologie sous-jacente et/ou des anomalies hématologiques concomitantes de la lignée plaquettaire et leucocytaire.
Les signes observés peuvent s'expliquer par l'hypoxie locale et l'effondrement de la barrière entre le sang et la rétine. Plus le taux d'hémoglobine est bas, plus l'atteinte est importante. La survenue de ces anomalies est plus fréquente en cas de thrombopénie associée.
Le degré de l'anémie peut s'apprécier par la couleur du pli conjonctival et par le reflet rouge pâle du fond d'œil [2].
On note l'aspect et la couleur porcelaine de la conjonctive, la pâleur du pli conjonctival.
On retrouve :
- –des hémorragies rétiniennes punctiformes ou en flammèches, si leur centre est de couleur blanche. Elles sont appelées taches de Roth (c'est un vaisseau rompu obturé par un thrombus de fibrine);
- –des nodules/exsudats cotonneux;
- –un aspect dilaté et tortueux des veines;
- –une opalescence de la rétine sensorielle;
- –un décollement exsudatif périphérique en cas d'anémie sévère;
- –une hémorragie maculaire : un cas d'hémorragie bilatérale a été décrit chez une jeune fille de 17 ans [3], dont les résultats hématologiques ont révélé une anémie mégalocytique, associée à une hémoglobine inférieure à 4,9 g/l. Aucune cause n'était associée;
- –un œdème : en cas de fortes pertes sanguines, un œdème papillaire et un œdème de la rétine peuvent être observés;
- –une neuropathie optique : des scotomes centrocæcaux peuvent apparaître en cas d'anémie pernicieuse (maladie de Biermer). De plus, l'anémie étant un facteur de risque d'hypertension intracrânienne, un œdème de stase peut s'observer.
Aksov et al. [4], dans leur étude portant sur la mesure de l'épaisseur des fibres nerveuses péripapillaires à l'OCT d'enfants atteints de thalassémie majeure ou de carence martiale, ont révélé que l'épaisseur des fibres était plus mince dans les quatre quadrants pour le groupe « thalassémie majeure » et dans le quadrant inférieur pour le groupe « carence martiale » .
L'épaisseur des fibres nerveuses ganglionnaires péripapillaires est corrélée au taux d'hémoglobine et au niveau de ferritine. Une autre étude [5] portant sur des enfants atteints de carence martiale a montré que l'épaisseur moyenne des fibres nerveuses péripapillaires dans les quadrants supérieur, inférieur et temporal était corrélée au taux d'hémoglobine.
La bêta-thalassémie majeure est une forme sévère et précoce de bêta-thalassémie caractérisée par une anémie nécessitant des transfusions régulières d'hématies. Parmi les nombreuses manifestations cliniques, des anomalies craniofaciales typiques à l'origine d'un retentissement sur les os orbitaires et la biométrie oculaire est possible. Nowroozzadek et al. [6] ont montré que les enfants atteints de bêta-thalassémie majeure avaient une longueur axiale plus petite, une courbure cornéenne modifiée associée à un astigmatisme irrégulier et un cristallin plus fin.
Cette pathologie associe une microcornée, une microphtalmie, un ptosis, des modifications de courbure cornéenne ainsi que des petites papilles [7]. La transmission est le plus souvent autosomique récessive. Les malformations congénitales associées, en particulier auditives, peuvent retarder les acquisitions visuelles. C'est pourquoi, il est important de réaliser très tôt un examen ophtalmologique, afin de dépister tout trouble visuel et de prescrire une correction adaptée. Ce point est corroboré par un cas clinique d'une enfant de 11 ans présentant un œil droit rouge et douloureux, avec effondrement de l'acuité visuelle dÛ à un glaucome néovasculaire. Ce dernier s'associait à une hémorragie intra-vitréenne, une néovascularisation papillaire et une rétinopathie ischémique périphérique. L'atteinte était bilatérale [8].
La drépanocytose, grande cause d'anémie chez l'enfant, est à l'origine de nombreuses manifestations oculaires (voir chapitre 16.2).
Aussi appelée microsphérocytose, cette maladie est de transmission autosomique dominante. L'ictère de cette anémie hémolytique congénitale peut atteindre les conjonctives, avec une teinte jaunâtre. Les autres manifestations ophtalmologiques possibles sont l'épicanthus, le ptosis, la vascularisation cornéenne limbique et un embryotoxon postérieur. La cataracte est souvent secondaire au traitement. Des malformations craniofaciales sont fréquentes, telles qu'un important écart interpupillaire, une microcéphalie, un rétrognatisme [9].
De transmission autosomique dominante, cette anémie peut s'associer à un épicanthus, un glaucome congénital et à un embryotoxon postérieur [9].
Cette anémie peut s'accompagner de baisse d'acuité visuelle, pâleur conjonctivale bilatérale et hémorragies maculaires prérétiniennes bilatérales. Les signes régressent après correction de la carence martiale [10].
Si les causes d'anémies chez l'enfant sont multiples, leurs manifestations oculaires sont peu spécifiques en dehors de la pâleur conjonctivale et des hémorragies au fond d'œil. L'atteinte ophtalmologique de l'anémie peut être associée à d'autres désordres hématologiques concomitants. Le traitement est avant tout celui de la cause.
[1] Bourrillon A, Benoist G, Delacourt C Pédiatrie:Elsevier Masson ( 2014 ) 551-556
[2] Tischendorf F, Meyer C, Spraul C Œil et médecine interne, modifications oculaires dans les maladies systémiques Les anomalies de la vision chez l'enfant et l'adolescent:Lavoisier ( 2005 ) 61-65
[3] Vaggu SK, Bhogadi P Bilateral macular hemorrhage due to megaloblastic anemia : a rare case report Indian J Ophthalmol: ( 2016 ) :64: 157-159
[4] Aksoy A, Aslan L, Aslankurt M Retinal fiber layer thickness in children with thalessemia major and iron deficiency anemia Semin Ophthalmol: ( 2014 ) :29: 22-26
[5] Türkyilmaz K, Oner V, Ozkasap S Peripapillary retinal nerve fiber layer thickness in children with iron deficiency anemia Eur J Ophthalmol: ( 2013 ) :23: 217-222
[6] Nowroozzadeh MH, Kalantari Z, Namvar K, Meshkibaf MH Ocular refractive and biometric characteristics in patients with thalassaemia major Clin Exp Optom: ( 2011 ) :94: 361-366
[7] Tornquist AL, Martin L, Winiarski J, Fahnehjelm KT Ocular manifestations and visual functions in patients with Fanconi anaemia Acta Ophthalmol: ( 2014 ) :92: 171-178
[8] Yahia SB, Touffahi SA, Zeghidi H Ocular neovascularization in a patient with Fanconi anemia Can J Ophthalmol: ( 2006 ) :41: 778-779
[9] Tsilou ET, Giri N, Weinstein S Ocular and orbital manifestations of the inherited bone marrow failure syndromes: Fanconi anemia and dyskeratosis congenita Ophthalmology: ( 2010 ) :117: 615-622
[10] Trivedi BP, Ravindran M, Pawar N Iron deficiency anemia presenting with macular star J AAPOS: ( 2015 ) :19: 486-487
S. Milazzo, O. Madar
Les cancers de l'enfant et de l'adolescent sont beaucoup plus rares que ceux de l'adulte; ils représentent moins de 1 % des cancers. Les leucémies aiguës sont les cancers les plus fréquents chez l'enfant avec un taux d'incidence de 30 % [1].
Ce sont des proliférations clonales malignes de précurseurs de cellules sanguines appelées blastes, bloqués à un stade précoce de leur différenciation.
On distingue deux sortes de leucémies aiguës :
- –les leucémies aiguës lymphoblastiques (LAL);
- –les leucémies aiguës myéloïdes (LAM);
Le type cytologique des blastes permet de différencier ces deux leucémies. Les LAL représentent 80 % des leucémies aiguës de l'enfant [1].
On compte environ 400 nouveaux cas par an en France, avec deux pics de fréquence :
- –de 2 à 5 ans;
- –à l'adolescence.
Les proliférations lymphoïdes immatures sont issues à 75 % de la lignée B et à 20 % de la lignée T [1].
- –Syndrome d'insuffisance médullaire : il correspond à l'atteinte des trois lignées (anémie, neutropénie, thrombopénie) et à leurs complications respectives.
- –Syndrome tumoral : il résulte de la prolifération et de l'accumulation de blastes dans le sang, la moelle osseuse, les organes hématopoïétiques et certains tissus de l'organisme.
- –Syndrome d'insuffisance médullaire : il correspond à l'atteinte des trois lignées (anémie, neutropénie, thrombopénie) et à leurs complications respectives.
- –Syndrome tumoral : il résulte de la prolifération et de l'accumulation de blastes dans le sang, la moelle osseuse, les organes hématopoïétiques et certains tissus de l'organisme.
L'atteinte oculaire est plus fréquente dans les formes aiguës que dans les formes chroniques. Les manifestations oculaires au cours des LAL sont plus fréquentes qu'au cours des LAM [2].
Presque toutes les structures oculaires peuvent être atteintes. Il faut néanmoins distinguer les atteintes primitives des atteintes secondaires.
Les atteintes primitives sont dues à une infiltration leucémique par invasion directe de cellules néoplasiques des tissus oculaires ou orbitaires. Il en est de même pour le système nerveux central, qui peut être envahi par des cellules blastiques.
Les atteintes secondaires peuvent être dues :
- –aux anomalies hématologiques associées telles que l'anémie, la thrombopénie;
- –aux complications de l'hyperviscosité;
- –à la survenue de maladies opportunistes due à l'immunodépression.
L'atteinte oculaire est donc fréquente, puisque présente dans 50 à 70 % des cas [3].
Les atteintes orbitaires peuvent révéler la maladie. Elles se manifestent par des troubles au niveau des muscles oculaires à l'origine d'ophtalmoplégie et de diplopie. Les signes cliniques comportent une exophtalmie, un œdème des paupières, un chémosis et des douleurs. Ces atteintes peuvent être uni- ou bilatérales. Elles sont souvent présentes à la phase terminale de la maladie. Elles touchent le plus souvent les parois latérales et médianes de l'orbite. L'atteinte orbitaire est un signe de mauvais pronostic [4].
Dans les LAM, la masse orbitaire correspond à un sarcome granulocytaire ou chlorome. Il faut éliminer les diagnostics différentiels tels le phlegmon de l'orbite et le rhabdomyosarcome. L'infiltration des voies lacrymales peut être à l'origine d'une dacryocystite.
Les signes cliniques sont l'œdème palpébral, un ptosis.
Les signes cliniques sont des hémorragies sous-conjonctivales spontanées, inhabituelles [5].
Un cas d'hémorragie sous-conjonctivale bilatérale massive révélant une LAM a été décrit chez un jeune homme de 20 ans [2].
Autres atteintes conjonctivales possibles : chémosis, infiltrats, anomalies vasculaires, conjonctivites.
Les signes cliniques sont des infiltrats limbiques, des ulcérations, une sécheresse, une abcédation.
Les cellules leucémiques peuvent envahir la chambre antérieure formant un Tyndall cellulaire, un pseudo-hypopion ou un hyphéma [5].
L'infiltration de l'angle par ces cellules peut être responsable d'un glaucome secondaire.
Le terme de « pseudo-uvéite » traduction de masquerade syndrome est utilisé pour caractériser ce type d'atteinte ressemblant à une uvéite mais ne répondant pas à un traitement anti-inflammatoire ou immunosuppresseur bien conduit.
Les signes cliniques sont un infiltrat leucémique, un épaississement de l'iris, des lésions nodulaires ou diffuses très rares, blanc laiteux avec hyperhémie intense. Souvent l'iris perd son architecture et devient hétérochromique. L'atteinte de l'iris est de pronostic défavorable et souvent en lien avec une atteinte méningée.
Les infiltrats choroïdiens sont plus fréquents que les infiltrats rétiniens.
Ils se manifestent par un épaississement jaune clair à rose, diffus, et peuvent se compliquer d'un décollement séreux rétinien, d'une altération atrophique ou hyperplasique de l'épithélium pigmentaire ou d'un épanchement ciliochoroïdien.
Ils sont parfois plus faciles à détecter à l'échographie, montrant un épaississement diffus de la choroïde [5].
On compte 28 à 50 % d'atteintes rétiniennes au cours des leucémies [6].
Les signes cliniques suivants sont relevés :
- –exsudats;
- –nodules cotonneux;
- –hémorragies intrarétiniennes et sous-hyaloïdiennes;
- –hémorragies rétiniennes dont le centre blanchâtre est formé de cellules leucémiques ou d'embols fibrinoplaquettaires, caractéristiques, appelées pseudo-taches de Roth, associées à d'autres hémorragies disséminées au pôle postérieur (fig. 16-31 a et b). Elles sont souvent bilatérales, localisées au pôle postérieur et en péripapillaire, elles peuvent toucher toutes les couches rétiniennes. Elles sont associées à une thrompocytopénie et/ou une anémie;
- –infiltrats leucémiques sous forme de dépôts jaunâtres dans la rétine et l'espace sous-rétinien;
- –infiltrats leucémiques périvasculaires produisant des stries gris blanchâtre au niveau de la rétine;
- –rarement, une atteinte du vitré résultant d'une extension via une hémorragie rétinienne;
- –syndrome d'hyperviscosité : une leucémie peut se compliquer d'un syndrome d'hyperviscosité avec ses atteintes ophtalmologiques (fig. 16-31c);
- –DR exsudatif [7].
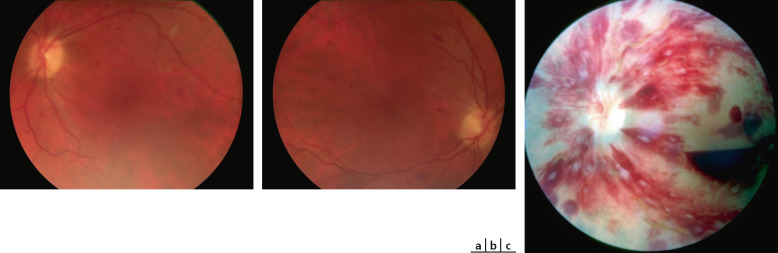
Fig. 16-31 Rétinographies.
a. OEil gauche : pseudo-taches de Roth associées à d’autres hémorragies disséminées. b. OEil droit : multiples hémorragies rétiniennes. c. OEil droit : syndrome d’hyperviscosité compliquant une leucémie aiguë : dilatation et tortuosité veineuse, taches de Roth chez une fillette de 7 ans.
Une infiltration leucémique du nerf optique se rencontre typiquement chez les enfants atteints de LAL. L'atteinte de la lame criblée s'accompagne d'un œdème de la papille. Lors d'une atteinte de la partie antérieure, on observe une hémorragie, un œdème de la papille. Lors d'une atteinte postérieure, on observe une hémorragie sur le disque optique.
Au total, l'œdème papillaire peut être secondaire à une infiltration primitive du nerf optique par des cellules blastiques, une infiltration des tissus orbitaires ou une hypertension intracrânienne.
L'atteinte infectieuse est souvent secondaire à la neutropénie ou au traitement immunosuppresseur. Elle peut se manifester par une rétinite, une uvéite postérieure, une endophtalmie ou un abcès cornéen. L'hypopion peut être le seul signe à révéler une LAM [8], même lors d'une rémission [9].
Sur ces terrains immunodéprimés, l'infection opportuniste peut se manifester, par exemple, par une choroïdite multifocale secondaire à une infection à Candida albicans [10].
Le principal traitement repose sur la chimiothérapie avec possibilité de greffe allogénique en cas de formes sévères à très sévères.
Pour les LAL de l'enfant, la rémission complète se situe aux alentours de 90 % et la guérison à 70 % [11].
Les principaux facteurs pronostiques favorable identifiés sont :
- –LAL : leucocytose basse, âge entre 1 et 10 ans, caractérisation biologique, réponse rapide au traitement;
- –LAM : caractérisation biologique.
L'examen de surveillance consiste à rechercher une sécheresse oculaire dans le cadre d'une maladie du greffon contre l'hôte, ainsi que des effets secondaires des chimiothérapies.
Les atteintes ophtalmologiques peuvent être révélatrices de la maladie [12, 13],
L'atteinte oculaire est associée à un mauvais pronostic vital si elle s'associe à des infiltrats rétiniens, une infiltration du nerf optique ou des hémorragies maculaires.
Elle est considérée par certains auteurs comme une atteinte du système nerveux central et nécessite un protocole thérapeutique particulier incluant une chimiothérapie intrathécale en association avec l'irradiation du système nerveux central et une intensification de la chimiothérapie systémique [2], L'examen ophtalmologique est obligatoire même en l'absence de symptôme [14, 15],
[1] Bourrillon A, Benoist G Cancers de l'enfant Pédiatrie:Elsevier Masson ( 2014 ) 580-589
[2] Taamallah-Malek I, Chebbi A, Bouladi M Hémorragie sous-conjonctivale bilatérale massive révélatrice d'une leucémie aiguë lymphoblastique J Fr Ophtalmol: ( 2013 ) :36: 45-48
[3] Tischendort FW, Meyer CH, Spraul CW, Dhem A Maladies hématologiques Œil et médecine interne, modifications oculaires dans les maladies systémiques:Lavoisier ( 2005 ) 61-66
[4] Ramamoorthy J, Jain R, Trehan A Orbital Mass in a child with acute lymphoblastic leukemia : a case report and review of the literature J Pediatr Hematol Oncol: ( 2016 ) :38: 646-648
[5] Kanski JJ, Milewski SA, Damato BE, Tanner V Pathologie vasculaire rétinienne Les pathologies du fond de l'œil – Atlas en ophtalmologie:Elsevier ( 2006 ) 85-86
[6] Dahredine M, El Moussaif H, Amazouzi A Hémorragie rétinienne à centre blanc révélant une leucémie aiguë lymphoblastique J Fr Ophtalmol: ( 2004 ) :27: 506-509
[7] Khaja WA, Pogrebniak AE, Bolling JP Combined orbital proptosis and exudative retinal detachment as initial manifestations of acute myeloidleukemia J AAPOS: ( 2015 ) :19: 479-482
[8] Alten F, Ehlert K, Böhm MR, Grenzebach UH Leukemic hypopyon in acute myeloid leukemia Eur J Ophthalmol: ( 2013 ) :23: 252-254
[9] Kulbacki E, Schneider E, Wang E ‘Hypopyon'in the anterior chamber : unilateral ocular relapse of acute myeloid leukaemia in a 2-year-old girl Br J Haematol: ( 2013 ) :162: 293-
[10] Santiagu F, Ong L, Ariffin WA, Tajunisah I A case of multifocal choroiditis secondary to Candida albicans infection in a leukemic child Ocul Immunol Inflamm: ( 2013 ) :21: 317-320
[11] Ifrah N, Cahn JY Hématologie. Collège des enseignants d'Hématologie, Société Française d'Hématologie:Elsevier Masson ( 2014 ) 63-74
[12] Chaudhry SR, Kreis AJ, Underhill HC, Madge SN Orbital mass secondary to acute lymphoblastic leukaemia in a child : a rare presentation Orbit: ( 2014 ) :33: 421-
[13] Chocron IM, Morrison DG, Friedman DL Ophthalmic manifestations of relapsing acute childhood leukemia J AAPOS: ( 2015 ) :19: 284-
[14] Bitirgen G, Belviranli S, Caliskan U Ophthalmic manifestations in recently diagnosed childhood leukemia Eur J Ophthalmol: ( 2016 ) :26: 88-91
[15] Khadka DN, Sharma AK, Shrestha GS Ocular manifestations of childhood acute leukemia in a tertiary level eye centre of Kathmandu, Nepal Nepal J Ophtalmol: ( 2014 ) :6: 197-204
S. Milazzo, O. Madar
Le syndrome d'hyperviscosité se définit comme l'ensemble des symptômes et manifestations cliniques secondaires à l'élévation de la viscosité sanguine ou plasmatique, entraînant un défaut d’oxygénation tissulaire [1, 2].
Les états d'hyperviscosité forment un groupe hétérogène de troubles rares secondaires à différentes perturbations cellulaires ou plasmatiques telles que :
- –une élévation de l'hématocrite (polyglobulie);
- –une élévation du nombre de leucocytes (hyperleucocytose massive) rencontrée notamment au cours des leucémies;
- –une élévation des protides ou de l'équilibre de ses constituants : en cas de syndrome inflammatoire ou dans les maladies telles que la maladie de Waldenstrom ou le myélome multiple des os;
- –une diminution de la déformabilité des hématies comme dans le cas particulier de la drépanocytose ou en cas d'acidose;
- –un accroissement de l'agrégation des hématies au cours des syndromes inflammatoires, des embolies graisseuses, de l'endotoxinémie.
Le syndrome d'hyperviscosité est une urgence thérapeutique médicale dont le diagnostic repose avant tout sur un faisceau d'arguments cliniques. Il doit être suspecté en présence de la triade suivante : saignements muqueux, troubles visuels, manifestations neurologiques [3]. Si la mesure de la viscosité sanguine ou plasmatique en permet le diagnostic de certitude à l'aide d'un viscosimètre, elle est rarement réalisée en pratique quotidienne [3].
Les étiologies diffèrent de celles de l'adulte, et sont essentiellement :
- –l'hyperviscosité plasmatique : c'est le cas des syndromes lymphoprolifératifs tels que la LAL, la LAM, le lymphome et le syndrome de Wiskott-Aldrich;
- –l'hyperviscosité cellulaire :
Il existe d'autres causes d'hyperviscosité : iatrogènes, cancers solides, syndrome inflammatoire, hypothermie, choc, brÛlure, transfusion massive.
Ces symptômes sont :
- –une baisse d'acuité visuelle;
- –une diplopie;
- –un tableau d'amaurose monoculaire transitoire expliqué par une hypoperfusion du globe oculaire responsable de cécité monoculaire transitoire ou bien d'une thrombose in situ d'une artère vascularisant le globe oculaire [4].
En cas de suspicion diagnostique, un fond d'œil doit être réalisé en urgence.
L'examen du segment antérieur est peu contributif mais il peut retrouver une tortuosité vasculaire conjonctivale.
Le fond d'œil constitue un élément indispensable lors de la prise en charge d'un syndrome d'hyperviscosité, tant pour le diagnostic que pour l'évaluation du pronostic et de l'efficacité du traitement.
Comme dit précédemment, l'atteinte ophtalmologique peut être liée à d'autres désordres hématologiques concomitants.
Les signes ophtalmologiques sont les suivants.
- –Hémorragies rétiniennes liées au ralentissement circulatoire et à l'ischémie rétinienne (fig. 16-32).
- –Néovascularisation rétinienne périphérique avec présence d'hémorragies rétiniennes périphériques. Dans les leucémies, un des signes évocateurs est l'existence d'hémorragie rétinienne centrée par un point blanc (taches de Roth). Dans les leucémies notamment aiguës, les hémorragies sont aussi liées à une thrombopathie aggravée par la thrombopénie.
- –Hémorragie intravitréenne non traumatique. Le traitement de l'hémorragie intravitréenne dense peut faire appel à la vitrectomie mais celle-ci est rendue délicate par les conditions d'hémostase locale et générale [5].
- –Anomalies vasculaires avec présence de vaisseaux dilatés, tortueux, secondaires à la stase sanguine (fig. 16-32) :
- –Atteintes de la microcirculation avec présence de nodules cotonneux, de micro-anévrismes.
- –Atteintes du nerf optique : neuropathie ischémique, œdème papillaire.
- –Œdème maculaire : secondaire au ralentissement circulatoire maculaire.
- –Décollement séreux rétinien ou décollement de l'épithélium pigmentaire.
L'atteinte au fond d'œil est commune à toutes les étiologies d'hyperviscosité et dépourvue de spécificité. Elle est caractérisée par une atteinte bilatérale, permettant ainsi la distinction avec son principal diagnostic différentiel : l'occlusion de la veine centrale de la rétine habituellement unilatérale.
Une régression rapide des lésions est habituellement observée avec la plasmaphérèse et le traitement étiologique.
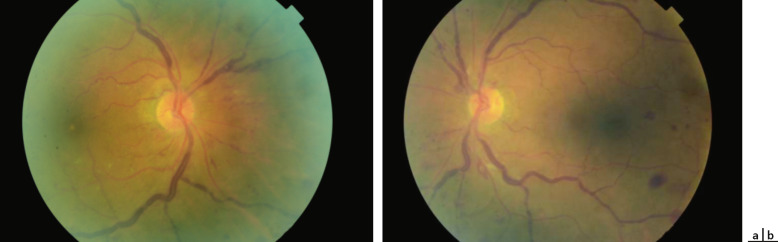
Fig. 16-32 Enfant de 15 ans atteint d’une leucémie myéloïde.
Rétinographies : dilatation et tortuosité veineuse, hémorragies rétiniennes disséminées de l’oeil droit (a) et de l’oeil gauche (b).
L'hyperviscosité sanguine locale est un facteur de risque vasculaire du glaucome en raison d'une moindre déformabilité érythrocytaire et/ou d'une hyperagrégabilité érythrocytaire. En effet, la réduction du flux sanguin oculaire est un facteur de risque d'incidence et de progression dans 50 % des glaucomes à pression normale [7].
Les cas suivants ont été décrits :
Quelle que soit sa cause, le syndrome d'hyperviscosité constitue une urgence thérapeutique [3]. L'objectif est une réduction rapide de la viscosité sanguine afin d'en contrôler les symptômes. Si le traitement étiologique est toujours nécessaire, il n'est généralement pas suffisant et certaines techniques spécialisées, comme la plasmaphérèse, s'imposent. Dans tous les cas, des mesures non spécifiques devront être rapidement mises en œuvre dès que le diagnostic est suspecté.
Il est impératif de maintenir un état d'hydratation, l'hémoconcentration majorant les troubles rhéologiques.
Un bilan biologique minimal (numération formule sanguine, VS, CRP) permet souvent de suspecter une hémopathie responsable avant un bilan hématologique ciblé.
Bien souvent, elle est déjà connue au moment du diagnostic, car des signes généraux sont présents tels que l'altération de l'état général, les céphalées, une épistaxis, des hémorragies digestives ou gynécologiques.
Bien que les signes ophtalmologiques soient rarement révélateurs de la maladie, il est essentiel de connaître les manifestations ophtalmologiques du syndrome d'hyperviscosité, car la prise en charge est urgente.
[1] Kanski JJ, Milewski SA, Damato BE, Tanner V Pathologie vasculaire rétinienne Les pathologies du fond de l'œil – Atlas en ophtalmologie:Elsevier ( 2006 ) 85-86
[2] Spalton DJ, Hitchings RA, Hunter PA, Tan JCH Rétine : affections vasculaires 1 Atlas d'ophtalmologie clinique:Elsevier ( 2005 ) 437-471
[3] Dumas G, Merceron S, Zafrani L Hyperviscosity syndrome Rev Med Interne: ( 2015 ) :36: 588-595
[4] Biousse V, Bousser MG Amaurose monoculaire transitoire. Encycl Méd Chir:Elsevier ( 1999 ) 1-10 Ophtalmologie, 21-490-A-20
[5] Ameline-Chalumeau B, Laroche A, Morel C, Larricart P :- Ophtalmologie, 21-248-A-26
[6] Rajagopal R, Apte RS Seeing through thick and through thin : retinal manifestations of thrombophilic and hyperviscositysyndromes Surv Ophthalmol: ( 2016 ) :61: 236-477
[7] Bresson-Dumont H, Hamard P Glaucome à pression normale. Encycl Méd Chir:Elsevier ( 2016 ) 1-9 Ophtalmologie, 21-280-B-45
[8] Jorge R, Scott IU, Oliveira RC Ocular findings in a patient with Castleman's disease before and after treatment with immunosuppression and plasmapheresis Ophthalmic Surg Lasers Imaging: ( 2010 ) :41: -
[9] Helal J, Malerbi FK, Melaragno Filho R Bilateral central retinal vein occlusion associated with blood hyperviscosity syndrome – case report Arq Bras Oftalmol: ( 2005 ) :68: 126-128
S. Milazzo, O. Madar
Les boucles vasculaires prépapillaires sont des anomalies vasculaires congénitales rares résultant d'anomalies de l'embryogenèse. Leur incidence dans la population générale est de 0,01 à 0,05 % [1]. Elles sont soit artérielles, soit veineuses, les artérielles étant les plus fréquentes. Elles sont le plus souvent unilatérales, asymptomatiques et de découverte fortuite. Ces boucles ne doivent pas être confondues avec une néovascularisation de la tête du nerf optique.
La vascularisation de l'œil au cours de son développement dépend principalement du système hyaloïdien qui est remplacé par les systèmes vasculaires définitifs à la naissance. Les boucles vasculaires prépapillaires sont des branches principales de l'artère ou de la veine centrale de la rétine qui quittent le disque optique en se dirigeant dans le vitré et en formant plusieurs boucles avant de retourner dans la papille. Parfois, les vaisseaux rétiniens en cours de développement progressent de façon erronée dans la cavité vitréenne par le canal de Cloquet au lieu de se diriger directement dans le plan rétinien. Ils décrivent alors une boucle dans le vitré avant de reprendre une configuration normale dans la rétine.
La boucle vasculaire prépapillaire correspond donc à des vaisseaux, semblant repliés sur eux-mêmes, qui émanent de la papille vers le vitré avec souvent un aspect en « tire-bouchon » . La boucle vasculaire relie deux branches de vaisseaux rétiniens définitifs. Un reliquat de la papille de Bergmeister peut être associé (fig. 16-33) [2]. Les boucles sont artérielles dans 95 % des cas [1] et peuvent alors présenter des pulsations. L'anomalie touche en général les vaisseaux inférieurs. C'est l'angiographie qui permet de faire la distinction entre une origine artérielle et une origine veineuse.
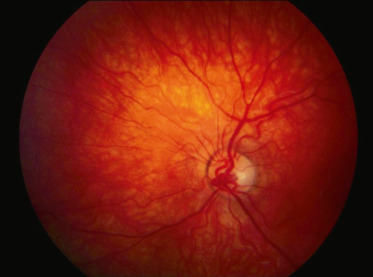
Fig. 16-33 Boucle vasculaire chez un enfant de 13 ans, de découverte fortuite.
Bien qu'elles soient en général asymptomatiques, les boucles artérielles peuvent parfois se compliquer de phénomènes ischémiques rétiniens dus soit à la thrombose de la boucle, soit à sa torsion, et à des phénomènes hémorragiques. La non-perfusion de la boucle, le retard de perfusion artérielle et la présence d'une artère cilior étinienne témoignent de l'origine artérielle de la boucle. La thrombose et la torsion sont les deux mécanismes connus de l'occlusion de la boucle. Le ralentissement circulatoire majeur dÛ à l'enroulement de la boucle expliquerait l'occlusion par torsion [3], Les complications [1] regroupent principalement la survenue de :
- –occlusion artérielle dans le territoire desservi par la branche rétinienne artérielle issue de la boucle;
- –amaurose fugace;
- –hémorragie intravitréenne;
- –hyphéma.
Les boucles veineuses sont beaucoup moins fréquentes. Aucune complication occlusive n'a été rapportée à ce jour.
Quelques cas de complications chez les enfants ont été publiés :
- –une occlusion de la branche inférieure de l'artère centrale de la rétine a été rapportée chez une jeune fille de 10 ans porteuse d'une boucle artérielle prépapillaire [4];
- –une occlusion d'artère rétinienne temporale inférieure chez un patient âgé de 16 ans porteur d'une boucle artérielle prépapillaire [5];
- –un cas d'occlusion de branche inférieure de l'artère centrale de la rétine a été décrit chez un adolescent après un traumatisme par collision lors d'un match de football [1]. Il s'agit toujours de cas isolés [6].
Kahn [7], à propos du suivi par angio-OCT d'un adolescent porteur d'une boucle vasculaire prépapillaire ayant présenté 2 ans auparavant une occlusion de branche de l'artère de la rétine secondaire à une torsion post-traumatique, précise l'intérêt de l'angio-OCT pour analyser le plexus superficiel et profond de la rétine, afin d'évaluer l'état de perfusion fovéolaire de manière non invasive.
Ces boucles ne doivent pas être confondues avec une néovascularisation de la tête du nerf optique. C'est préciser l'importance d'examiner tout le champ rétinien, en particulier sa périphérie.
Les boucles vasculaires prépapillaires sont des anomalies vasculaires congénitales rares, l'incidence dans la population générale étant de 0,01 à 0,05 % . Elles sont le plus souvent unilatérales et asymptomatiques. Elles peuvent se compliquer de phénomènes ischémiques par torsion ou thrombose de la boucle. Ces complications surviennent plus fréquemment chez l'adulte mais quelques cas d'occlusion de branche inférieure de l'artère centrale de la rétine compliquant une boucle artérielle prépapillaire ont été publiés chez l'enfant.
[1] Rahimy E, Rayess N, Talamini CL, Kaiser RS Traumatic prepapillary loop torsion and associated branch retinal artery occlusion JAMA Ophthalmol: ( 2014 ) :132: 1376-1377
[2] Shakin EP, Shields JA, Augsburger JJ, Brown GC Clinico-pathologic correlation of a prepapillary vascular loop Retina: ( 1988 ) :8: 55-
[3] Zekraoui Y, Nafizy I, Hajji Z Boucle vasculaire prépapillaire bilatérale compliquée d'une occlusion artérielle J Fr Ophtalmol: ( 2010 ) :33: 715-717
[4] Singh R, Fujinami K, Moore AT Branch retinal artery occlusion secondary to prepapillary arterial loop Retin Cases Brief Rep: ( 2014 ) :8: 124-126
[5] Bonneric JG, Boissonnot M, Rovira JC, Dighiero P Une boucle artérielle prépapillaire compliquée d'occlusion de branche artérielle rétinienne J Fr Ophtalmol: ( 2007 ) :30: e8-
[6] Delyfer MN, Rougier MB, François L, Korobelnik JF Occlusion of a retinal branch in two young patients presenting a congenital prepapillary vascular loop J Fr Ophtalmol: ( 2008 ) :31: 126-127
[7] Khan MA, Shahlaee A, Kaiser RS Two-year follow-up of a prepapillary vascular looprelated branch retinal artery occlusion JAMA Ophthalmol: ( 2016 ) :134: e153670-
- Chapitre 16
Pathologie vasculaire- 1 – Rétinopathie des prématurés
- 2 – Syndromes drépanocytaires
- 3 – Maladie de Coats
- 4 – Dystrophie facio-scapulo-humérale chez l’enfant
- 5 – Diabète et atteintes rétiniennes chez l’enfant
- 6 – Rétinopathie hypertensive
- 7 – Maladie de Eales en pédiatrie
- 8 – Incontinentia pigmenti
- 9 – OEil et syndrome de Goodpasture
- 10 – Syndrome de Churg et Strauss
- 11 – Artérite de Takayasu
- 12 – Atteintes ophtalmologiques de l’anémie en pédiatrie
- 13 – Leucémie et atteinte ophtalmologique en pédiatrie
- 14 – Syndrome d’hyperviscosité et atteinte ophtalmologique pédiatrique
- 15 – Boucle vasculaire prépapillaire en pédiatrie