Pathologie neuro-ophtalmologique d’origine cérébrale
Coordonné par S. Defoort-Dhellemmes
S. Defoort-Dhellemmes
Ce chapitre concerne les atteintes des voies visuelles d’origine cérébrale qui constituent la première cause de cécité et de malvoyance profonde dans les pays à haut revenu (voir chapitre 1). Ces pathologies cérébrales sont présentées par ordre d’apparition. Ce sont les malformations dues à des anomalies du 1er trimestre de grossesse, les lésions périnatales puis les pathologies de l’enfance et l’adolescence. Certaines sont associées à un polyhandicap, nécessitant une prise en charge précoce. Le mode de prise en charge et les techniques d’examens réalisés dépendent de l’âge de l’enfant.
Les enfants atteints de lésions cérébrales, qu’ils soient nés prématurément ou à terme, sont à haut risque de troubles visuels. Ils doivent donc être vus par un ophtalmologiste à la naissance et il est recommandé un examen ophtalmologique entre l’âge de 3 et 12 mois avec un fond d’oeil et une réfraction sous cycloplégie même en l’absence de signes d’appel [1].
[1] Speeg-Schatz C, Geffrier d’Acremont C. Dépistage précoce des troubles de la fonction visuelle chez l’enfant pour prévenir l’amblyopie. Paris : ANAES ; octobre 2002.
I . Bouvet-Drumare
Cette malformation est marquée par une anomalie de la position de la jonction bulbomédullaire et du cervelet, sous le trou occipital. Selon la position de cette jonction et les anomalies associées on en décrit trois types (I, II, III) dont essentiellement les types I et II concernent l’enfant :
type I : ptose des amygdales cérébelleuses à 5 mm au moins sous le trou occipital, associée à une syringomyélie dans 10 à 15 % des cas ;
type II : descente du tronc cérébral et du vermis cérébelleux dans le canal médullaire cervical, avec abaissement de la tente du cervelet, associée à une myéloméningocèle sacrée dans 95 % des cas.
Strabisme concomitant aigu, nystagmus (essentiellement nystagmus battant vers le bas qui génère des oscillopsies et une baisse d’acuité visuelle de près, hautement évocateur chez l’enfant, ou nystagmus horizontal, alternant périodique), oedème papillaire secondaire à l’hydrocéphalie.
Essentiellement céphalées et douleurs cervicales dans le type I, plus rarement ataxie, vertiges, paralysie des dernières paires crâniennes, déficit des extrémités (mains et pieds). Dans le type II, dès la naissance, troubles respiratoires pouvant conduire au décès, hypotonie, troubles sphinctériens, troubles de la succion et de la déglutition.
L’imagerie par résonance magnétique (IRM) cérébromédullaire et sagittale est l’examen essentiel au diagnostic ; dans le type II, le diagnostic est fréquemment fait en anténatal par l’échographie et l’IRM.
Le traitement est neurochirurgical. Il ne concerne que les formes symptomatiques dans le type I, alors qu’il est obligatoire et plus complexe pour le type II. La prise en charge chirurgicale du strabisme ou du nystagmus n’est indiquée qu’après le traitement neurochirurgical compte tenu des améliorations importantes des signes ophtalmologiques après la neurochirurgie (fig. 22-1).
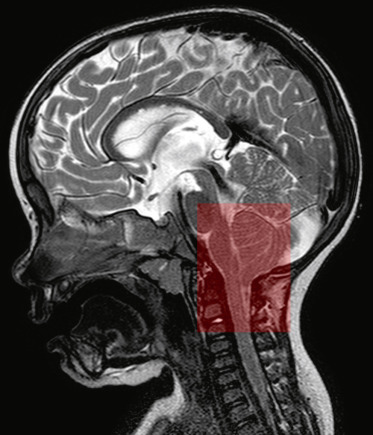
Fig. 22-1 Garçon âgé de 5 mois adressé pour mauvais suivi visuel et nystagmus congénital horizontal
LAF et FO normaux. PEV et ERG normaux. IRM cérébrale : malformation de Chiari type I qui évolue progressivement vers un type II. (Le caractère horizontal pendulaire d’un nystagmus est atypique dans ce type de malformation.)
Cette malformation cérébrale complexe est due à un défaut de clivage médian du prosencéphale, survenant entre le 18e et le 28e jour de gestation, touchant le cerveau antérieur et le visage, à l’origine de manifestations neurologiques et d’anomalies faciales de degré variable. Elle est fréquente (1/10 000 naissances, 1/250 produits de conception).
On retrouve : cyclopie, synophtalmie, hypotélorisme, colobome choriorétinien, amétropie, microcornée, chambre antérieure étroite, strabisme, dysplasie rétinienne, hypoplasie ou aplasie des nerfs optiques.
On retrouve : agénésie prémaxillaire, fente médiane ou bilatérale labiale et/ou palatine, atrésie des choanes, sténose du sinus pyriforme, hypotélorisme et incisive maxillaire médiane unique ; la face peut être normale.
Le diagnostic repose sur l’IRM et le diagnostic moléculaire – 14 gènes impliqués dont quatre majeurs : SHH (7q36), ZIC2 (13q32),SIX3 (2p21), TGIF (18p11). Le diagnostic prénatal repose essentiellement sur l’échographie et l’IRM.
Le pronostic est variable selon la sévérité de la malformation ; le pronostic vital est engagé et chez les enfants qui survivent, de nombreuses manifestations associées sont décrites telles que : retard du développement, hydrocéphalie, déficit moteur, troubles d’alimentation et de déglutition, épilepsie, dysrégulation hypothalamique, troubles endocriniens d’origine hypophysaire (diabète insipide). Il existe des formes familiales avec une forte variabilité de phénotype intrafamiliale (fig. 22-2 et 22-3).
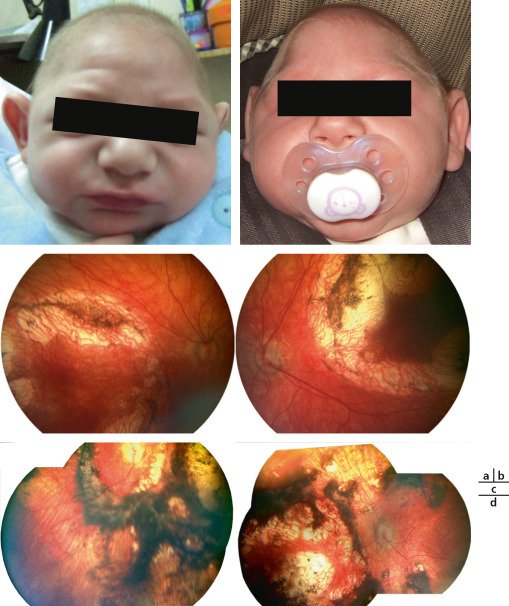
Fig. 22-2 Jumeaux (a, b) atteints d’holoprosencéphalie.
Jumeau en a, FO : lacunes choriorétiniennes au pôle postérieur (c). Jumeau en b, FO : lacunes choriorétiniennes beaucoup plus étendues (d).
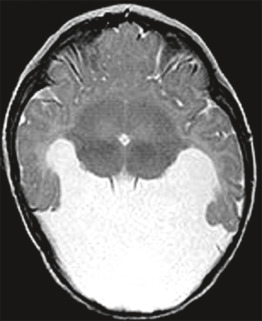
Fig. 22-3 IRM holoprosencéphalie (fusion diencéphale et lobes frontaux, ventricule unique)
Remerciements au Dr G. Soto Ares.)
Cette entité clinique hétérogène se définit par la triade classique : hypoplasie des nerfs optiques, déficits hormonaux hypophysaires et malformations cérébrales de la ligne médiane. Elle est également connue sous le nom de syndrome de De Morsier. Son incidence est estimée à 1/10 000 naissances vivantes. Sa sévérité est variable, la triade complète n’est présente que chez 30 % des patients, mais au moins deux des trois signes doivent être présents pour poser le diagnostic.
La dysplasie septo-optique est souvent découverte devant des signes de malvoyance chez un nourrisson (pas de suivi visuel, comportement de cécité), ou devant un nystagmus infantile. Si les nerfs optiques ne sont pas atrophiques ou si l’hypoplasie est unilatérale le comportement visuel peut être normal, et être associé à un strabisme sensoriel (formes unilatérales, avec parfois syndrome du monophtalme fonctionnel). L’examen en lampe à fente (LAF) est normal ; les potentiels évoqués visuels (PEV) en stimulation par flashes, s’ils sont enregistrés, sont plats ou déstructurés uniou bilatéralement, traduisant la sévérité de l’atteinte des voies optiques. S’ils sont normaux ou validés, le pronostic visuel est meilleur. Le diagnostic est fait au fond d’oeil (FO) : les papilles sont de petite taille, l’émergence des vaisseaux se fait au centre de la papille et elles sont cernées d’un halo hypo- ou hyperpigmenté
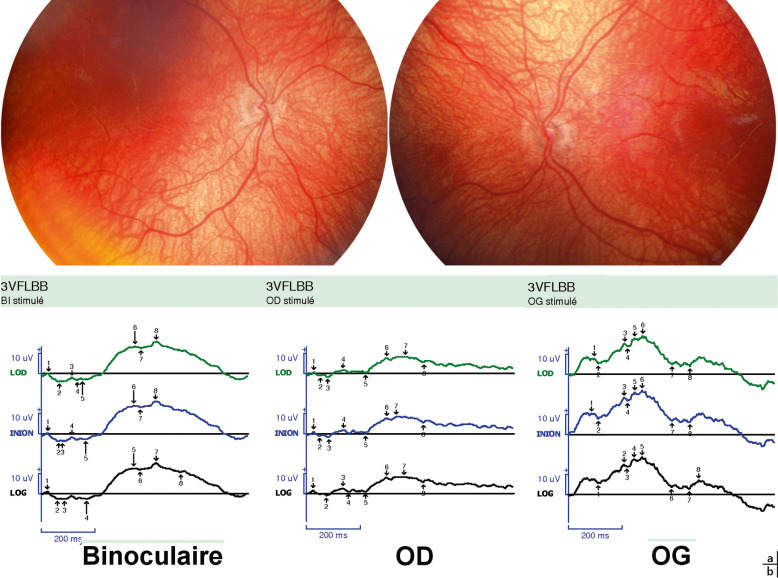
Fig. 22-4 Enfant de 2 mois et demi adressé pour comportement de cécité.
a. FO : hypoplasie sévère des nerfs optiques avec atrophie papillaire, atrophie péripapillaire (double anneau). b. PEV : présence uniquement d’une réponse en stimulation par flashes en binoculaire sous forme d’une onde tardive (360 ms) et immature.
réalisant l’aspect du « double anneau » . Elles peuvent être confondues avec une excavation papillaire centrale (fig. 22-4).
L’IRM cérébrale confirme le diagnostic en révélant : l’agénésie du septum pellucidum qui n’est ni constante ni pathognomonique, une hypoplasie du corps calleux, ou d’autres anomalies non spécifiques (hypoplasie de la substance blanche, hétérotopie corticale, polymicrogyrie, schizencéphalie, pachygyrie, kystes arachnoïdiens). Surtout elle précise l’atteinte de la tige pituitaire et de l’antéhypophyse (fig. 22-5).
Le bilan est toujours complété par un examen endocrinien même dans les formes unilatérales, à la recherche d’un déficit en hormone de croissance (70 % des cas), d’un hypothyroïdisme (43 % ), d’un déficit en adenocorticotrophic hormone (ACTH) (27 % ) et d’un diabète insipide (2 % ) ; s’il est normal, il faudra surveiller la courbe staturopondérale jusqu’à l’âge de la puberté et redoser l’hormone de croissance en cas de fléchissement de celle-ci, certains déficits partiels ne se révélant que plus tard (formes unilatérales).
Il est nécessaire d’adresser les patients sévèrement malvoyants vers des structures spécialisées dans la déficience visuelle très rapidement pour éviter les complications neuropsychologiques liées à celle-ci
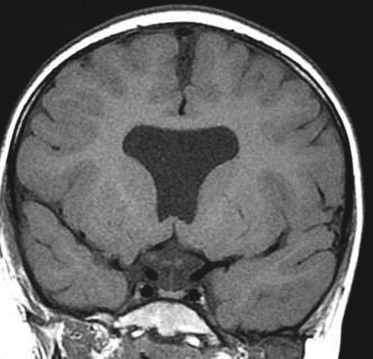
Fig. 22-5 Hypoplasie du chiasma optique, pas de septum pellucidum = dysplasie septo-optique.
(Remerciements au Dr G. Soto Ares.)
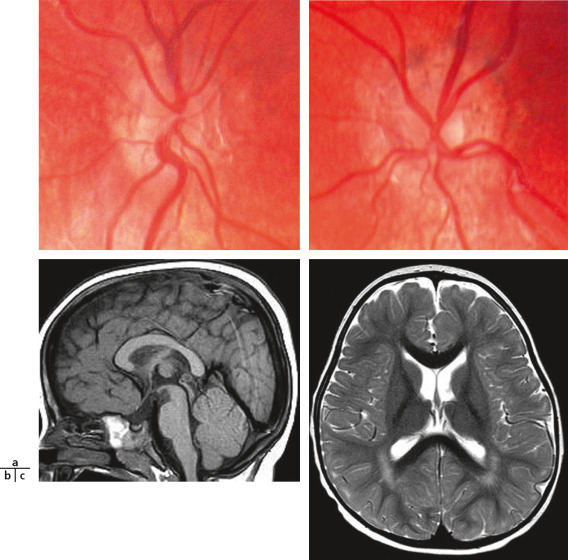
Fig. 22-6 Enfant de 1 an adressé pour strabisme convergent gauche, amblyopie gauche et nystagmus horizontal.
a. FO : hypoplasie bilatérale des nerfs optiques, discrète sur l’oeil droit et nette sur l’oeil gauche. b, c. IRM, séquence 3D Drive : hypoplasie du chiasma optique asymétrique, plus sévère à gauche (b) ; pas de visibilité de la tige pituitaire ; intégrité du septum pellucidum (c) ; pas de dysplasie septo-optique. (fig. 22-6b et c : remerciements au Dr G. Soto Ares.)
(retard de développement, troubles de type autistique). Dans le cas de formes mineures, avec atteinte visuelle moins sévère, le diagnostic est parfois fait tard et les patients mènent une vie normale.
La majorité des cas sont sporadiques. Le facteur de risque le plus retrouvé dans toutes les études est le jeune âge maternel. L’exposition à des drogues (LSD, traitements anticonvulsivants, antidépresseurs, quinine ou alcool) est moins certaine. Plusieurs gènes sont en cause : HESX1 (3p21.2-p21.1), soit homozygotes (autosomique récessive [AR]), soit hétérozygotes (autosomique dominante [AD]) ; SOX2 (3q26.3-q27) ; des mutations/duplications de SOX3 (Xq26.3) ; des mutations d’OTX2 (14q21-q22. Une mutation n’est retrouvée que chez moins de 1 % des patients (fig. 22-6)
Ces anomalies sont beaucoup mieux connues depuis l’apport de l’IRM ; elles peuvent être dues à des mutations génétiques ou à l’exposition à des toxiques (alcool, drogues).
Ce trouble se traduit essentiellement par des microcéphalies.
Les pathologies associant une microcéphalie syndromique sont nombreuses. Pour l’ophtalmologiste, nous retiendrons l’association microcéphalie et choriorétinopathie : il s’agit d’une microcéphalie vraie avec retard mental modéré et retard de croissance, associée à une dystrophie rétinienne bâtonnets-cônes. Au FO à la naissance, on note des lacunes choriorétiniennes sous l’arcade
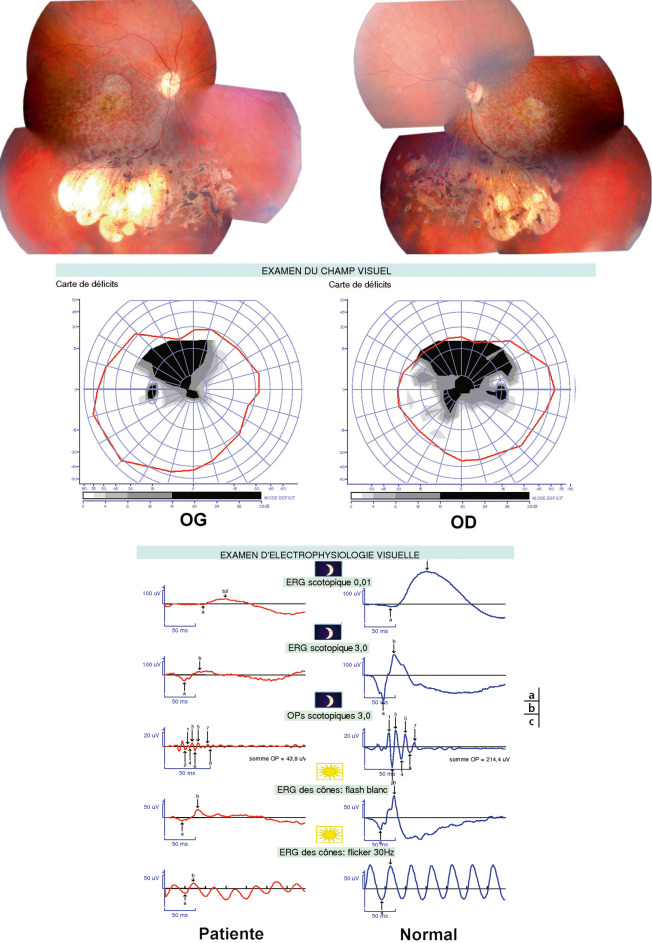
Fig. 22-7 Jeune fille âgée de 24 ans présentant une microcéphalie et des lacunes choriorétiniennes depuis la naissance.
(sa mère est atteinte). Disposition des lacunes en inférieur, sous l’arcade des vaisseaux temporaux, bilatérale, caractéristique (syndrome d’Alzial-Dufier). b. Atteinte du champ visuel qui correspond à la disposition des lacunes (noir = non vu). c. Atteinte de l’ERG qui est très diminué en scotopique et en photopique = dystrophie rétinienne bâtonnets–cônes.
des vaisseaux temporaux inférieurs, associées à une rétinopathie avec atteinte des composantes scotopiques et photopiques de l’électrorétinogramme (ERG), évoluant progressivement comme une dystrophie bâtonnets-cônes. Il existe des porteurs sains ou des patients avec pénétrance incomplète (fig. 22-7). Le diagnostic différentiel est le syndrome d’Aicardi, qui ne touche que les filles et associe des spasmes en flexion à un retard intellectuel sévère, avec des lacunes choriorétiniennes sans dystrophie rétinienne à l’ERG (voir chapitre 21.1).
- Hétérotopies nodulaires périventriculaires : elles sont constituées par un amas de neurones n’ayant pas effectué leur migration et se trouvant ainsi en dehors du cortex, le plus souvent au niveau périventriculaire. Elles peuvent être syndromiques comme dans le syndrome d’Aicardi (voir chapitre 16.1) ou le syndrome de Zellweger (peroxysomopathie caractérisée par une dysmorphie faciale très caractéristique, une hypotonie sévère, des crises d’épilepsie et des dysfonctionnements hépatiques et rénaux). Les patients peuvent également présenter une cataracte, un glaucome, une dystrophie rétinienne mixte, un nystagmus et une atrophie du nerf optique. Progressivement surviennent des troubles visuels puis la perte de la vision. Une surdité neurosensorielle peut être présente souvent secondaire à un défaut génétique du gène PEX1.
- Hétérotopies en bande sous-corticales : elles sont beaucoup moins graves et ne donnent qu’un retard mental léger. Il n’y a pas de signes ophtalmologiques spécifiques.
- Lissencéphalies : elles sont caractérisées par une surface cérébrale lisse ou avec très peu de sillons. Elles sont responsables d’une épilepsie sévère, de troubles du tonus et d’un retard mental important. Il n’y a pas de signes ophtalmologiques spécifiques.
- Polymicrogyrie : anomalie locale du cortex cérébral qui se caractérise par la présence d’un excès de giration et de sillons peu profonds. Elle est responsable d’une épilepsie sévère et d’un retard mental modéré. Elle peut être secondaire à une infection intra-utérine à cytomégalovirus et on peut alors retrouver au FO des cicatrices rétiniennes ou une atrophie optique. Elle peut être associée également au syndrome de Zellweger.
- Schizencéphalie : elle se caractérise par une fente linéaire bordée de substance grise traversant tout l’hémisphère du ventricule. Elle entraîne des troubles moteurs, une épilepsie et un retard psychomoteur à des degrés divers.
Anomalie la plus fréquente, elle associe une hypoplasie du vermis et une dilatation kystique du V4 éloignant le vermis du tronc cérébral. Elle entraîne une hydrocéphalie, une encéphalocèle, une macrocéphalie et un retard de développement psychomoteur. Au FO, on peut avoir initialement un oedème papillaire, puis une atrophie optique et un aspect de pseudocolobome maculaire.
Ce syndrome rassemble un ensemble de pathologies qui ont en commun une agénésie ou dysgénésie du vermis cérébelleux se manifestant par le signe pathognomonique de la « dent molaire » sur les coupes axiales de l’IRM au niveau de la jonction pontomésencéphalique. Sur les coupes sagittales, cette hypodysplasie ou dysplasie s’accompagne d’une horizontalisation du V4 (fig. 22-8) [4].
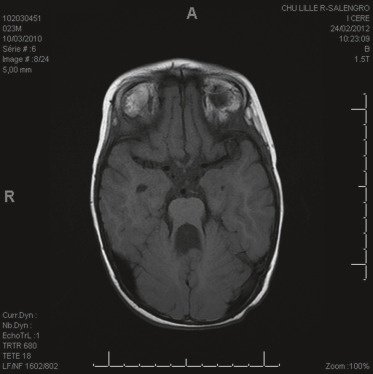
Fig. 22-8 Aspect IRM du syndrome de Joubert : hypoplasie du vermis cérébelleux donnant un aspect de dent molaire.
Chez le nourrisson, le syndrome de Joubert associe des troubles respiratoires, une hypotonie et fréquemment des mouvements oculomoteurs anormaux en particulier un nystagmus. Puis apparaissent une ataxie qui perturbe l’apprentissage de la marche, une apraxie oculomotrice, un retard de développement psychomoteur. Les facultés intellectuelles peuvent être normales ou gravement altérées. Ces signes neurologiques peuvent être isolés ou associés à une atteinte rénale, hépatique, osseuse (scoliose, dysplasie osseuse), cardiaque, malformative (face et polydactylie).
L’apraxie oculomotrice est presque constante, le nystagmus est infantile précoce et nécessite de faire un ERG pour rechercher une dystrophie rétinienne. Il s’agit d’une dystrophie mixte, soit majeure (amaurose congénitale de Leber), soit précoce et progressive (dystrophie rétinienne d’apparition précoce dont on sait qu’elle forme un continuum de pathologies entre l’amaurose de Leber et les dystrophies rétiniennes mixtes de l’enfance). Le FO peut être normal ou retrouver des modifications de pigmentation évoquant une atteinte rétinienne. Le FO peut aussi retrouver un colobome choriorétinien uni- ou bilatéral. Les formes avec colobome sont plus souvent associées au syndrome de Joubert avec atteinte hépatique. Les formes avec dystrophie rétinienne peuvent être également associées à une atteinte rénale (syndrome de Senior-Loken) (fig. 22-9).
Il existe actuellement 21 gènes qui codent tous pour des protéines du cil primitif, ce qui fait du syndrome de Joubert une ciliopathie. La transmission est autosomale récessive ou récessive liée à l’X (plus rare). Pour certaines formes de la maladie, il existe des corrélations génotype-phénotype intéressantes telles que l’association du gène CEP290 aux formes cérébro-oculo-rénales avec dystrophie rétinienne type amaurose congénitale de Leber et néphro nophtise ou l’association du gène TMEM67 avec une atteinte hépatique. Néanmoins plusieurs gènes peuvent être responsables du syndrome de Joubert avec dystrophie rétinienne (AHI1, INPP5E, ARL13B, CC2D2A, NPHP1, RPGRIP1L et TMEM237). L’intérêt des corrélations génotype-phénotype est de guider la recherche des affections associées et la surveillance. Ce groupe d’affections est accessible au diagnostic anténatal, et le dépistage peut surtout se faire au moyen de l’échographie anténatale et de l’IRM dans les familles exposées.
Pour l’ophtalmologiste, il s’agit d’évaluer la présence d’une déficience visuelle et sa sévérité pour :
- proposer l’aide d’un réseau de soins si nécessaire : centre d’action médico-social précoce (CAMPS) déficient visuel, puis service d’accompagnement à l’acquisition de l’autonomie et à la scolarisation (SAAAS) ;
- proposer une photoprotection en cas de dystrophie rétinienne ;
- organiser une rééducation orthoptique de l’apraxie oculomotrice chaque fois que la coopération de l’enfant le permet.
On en rapproche les anomalies trouvées dans le syndrome PHACES pour Posterior fossa malformations, Hemangiomas, Arterial anomalies, Cardiac defects and coarctation of the Aorta, Eye abnormalities, and Sternal abnormalities or ventral developmental defects (fig. 22-10). Ce syndrome est sporadique et touche des filles dans 90 % des cas. Les anomalies oculaires sont variées : glaucome congénital, hyperplasie du vitré primitif et hypoplasie des nerfs optiques, colobomes, microphtalmie et cryptophtalmie. Il peut exister un hémangiome de la face, à différencier de l’angiome plan du syndrome de Sturge-Weber-Krabbe.
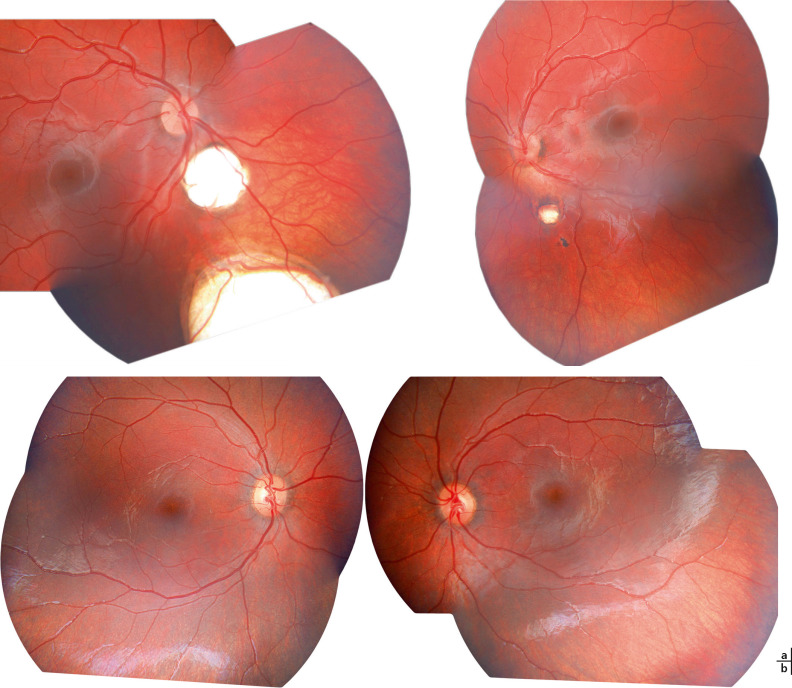
Fig. 22-9 Syndrome de Joubert.
a. Diagnostiqué sur une IRM motivée par un retard d’acquisitions (enfant de 20 mois), avec colobome choriorétinien bilatéral. b. Avec dystrophie rétinienne : modification de la pigmentation en périphérie.
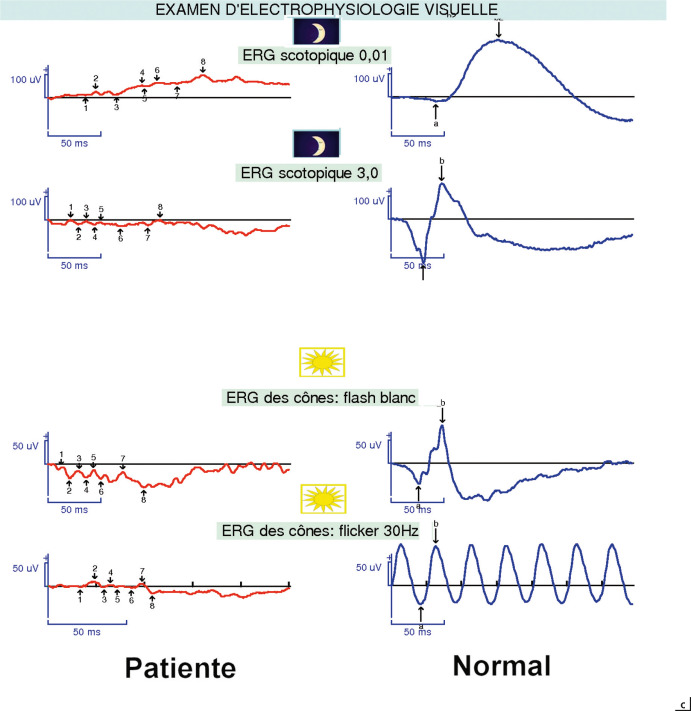
Fig. 22-9 Syndrome de Joubert. (Suite)
c. Avec dystrophie rétinienne : ERG altéré en scotopique et en photopique.
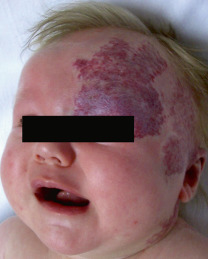
Fig. 22-10 Enfant atteinte de syndrome de PHACES.
Les hypoplasies pontocérébelleuses se traduisent par une diminution sévère de la taille du cervelet et du tronc cérébral congénitale ; il en existe sept types. Dans le type 3, il existe une atrophie optique.
Cette malformation associe une paralysie complète des saccades horizontales avec parfois un nystagmus battant vers le haut, une scoliose progressive [5]. À l’IRM, on note une dilatation du V4 et un aspect en papillon du bulbe sur les coupes axiales. Des mutations du gène ROBO3 (11q23-q25) ont été retrouvées.
Ce syndrome associe une diplégie faciale, complète dans 30 % des cas et bilatérale dans 92 % des cas, à une paralysie du VI dans 80 % des cas en règle bilatérale (avec impossibilité d’abduction au-delà de la ligne médiane), responsable d’un strabisme convergent bilatéral avec, souvent, fixation croisée. Ces anomalies sont responsables d’une pauvreté de l’expression du visage (amimie ou hypomimie) caractéristique.
Les malformations anténatales du système nerveux central retentissent sur le développement des voies optiques et de l’oeil. Elles peuvent être découvertes au décours d’un examen ophtalmologique. Il faut retenir que :
la malformation de Chiari se manifeste par des troubles oculomoteurs (en particulier nystagmus battant vers le bas) et elle nécessite un avis neurochirurgical du fait du risque de décompensation ;
les hypoplasies de la papille, uni- ou bilatérales, nécessitent une imagerie et un bilan endocrinien à la recherche d’un syndrome de De Morsier ;
toute apraxie oculomotrice nécessite un bilan étiologique pour éliminer un syndrome de Joubert, qui peut donner une dystrophie rétinienne mixte ou un colobome sous-papillaire et de nombreuses autres atteintes viscérales ;
dans le cas d’une microcéphalie, il faut rechercher des lacunes choriorétiniennes et une dystrophie rétinienne.
L’examen clinique recherche d’autres signes neurologiques associés : des troubles de la déglutition et de la ventilation, une limitation d’ouverture de la bouche, une voix nasonnée, parfois une déficience auditive, ou des anomalies neurologiques générales
Il existe également des malformations faciales (palais ogival, épicanthus, hypertélorisme, micrognathie, anomalies des lobes auriculaires ou des dents, etc.), une agénésie musculaire (syndrome de Poland), des anomalies des extrémités des membres et en particulier un pied bot varus équin, voire des malformations cardiaques ou cérébrales.
Sa genèse n’est pas certaine mais il est probablement dû à un accident vasculaire du tronc cérébral pendant la période embryonnaire. Il existe cependant des formes héréditaires à transmission autosomique dominante.
Le diagnostic différentiel doit éliminer une myasthénie du nourrisson ou une myopathie.
L’IRM cérébrale est le plus souvent normale.
La prise en charge dépend de l’intensité des symptômes : chez le petit enfant, elle concernera essentiellement les troubles de la succion, de la déglutition et de l’alimentation ainsi que le pied bot.
L’ophtalmologiste cherchera à dépister et à traiter l’amblyopie puis à traiter chirurgicalement le strabisme et éventuellement le ptosis.
[1] Grill J, Roujeau T, Bouchireb K, et al. Malformations cérébrales, médullaires et crâniennes. Neurologie pédiatrique. Médecine Sciences Flammarion ; 2010.
[2] Pineda-Alvarez DE, Solomon BD, Roessler E et al. A broad range of ophthalmologic anomalies is part of the holoprosencephaly spectrum. Am J Med Genet A 2011 ; 155A 2713-20.
[3] Garcia-Filion P, Borchert M. Optic nerve hypoplasia syndrome : a review of the epidemiology and clinical associations. Curr Treat Options Neurol 2013 ; 15 : 78-89.
[4] Romani M, Micalizzi A, Valente EM. Joubert syndrome : congenital cerebellar ataxia with the molar tooth. Lancet Neurol 2013 ; 12 : 894-905.
[5] Samoladas EP, O’Dowd J, Cardoso-Almeida A, Demetriades AK. Horizontal gaze palsy and scoliosis : a case report and review of the literature. Hippokratia 2013 ; 17 : 370-2.
I. Bouacha
Les lésions cérébrales périnatales sont responsables de manifestations ophtalmologiques qui vont soit aggraver le pronostic fonctionnel d’enfants déjà lourdement handicapés, soit constituer à terme le trouble principal dont les symptômes ne seront démasqués qu’à l’âge scolaire. La période périnatale est diversement appréciée dans les différents pays. Nous retiendrons la définition de l’Organisation mondiale de la santé (OMS) qui la définit comme une période qui débute à la 22e semaine d’aménorrhée (SA ; 154 jours) et se termine 7 jours révolus après la naissance. Cette approche élimine les malformations chromosomiques ou génétiques, les embryofoetopathies infectieuses de survenue précoce, antérieure à cette période périnatale, et les lésions infectieuses, métaboliques, traumatiques ou tumorales qui, elles, sont postérieures à cette période.
Les lésions cérébrales périnatales sont conditionnées par l’âge de survenue de l’atteinte, sa durée et sa sévérité [1]. Ce sont : les lésions d’anoxie ou hypoxie-ischémie et les hémorragies intracérébrales dont la localisation est différente chez le nouveau-né à terme et le prématuré ; les traumatismes obstétricaux ; la leucomalacie périventriculaire (LMPV) qui est l’apanage du prématuré.
L’anoxie périnatale affecte 2 à 4 ‰ des naissances à terme, entraînant une encéphalopathie anoxo-ischémique de gravité variable. Les régions les plus touchées sont la substance grise corticale, les noyaux gris centraux et la substance blanche sous-corticale. Les hémorragies intracérébrales, de siège intraventriculaire, sont souvent associées à l’asphyxie et aux désordres métaboliques. Les traumatismes obstétricaux peuvent être à l’origine de paralysies oculomotrices.
Les lésions hypoxo-ischémiques touchent principalement la substance blanche périventriculaire immature, aboutissant à la LMPV. Les hémorragies cérébrales de siège épendymaire et intraventriculaire surviennent essentiellement chez le nouveau-né de moins de 32 SA. Asymétriques et de gravité variable, elles ont pour principales complications l’hydrocéphalie post-hémorragique et la LMPV. Les traumatismes obstétricaux sont nettement moins fréquents que chez le nouveau-né à terme. La leucomalacie périventriculaire augmente de fréquence avec le degré de prématurité. Les lésions sont habituellement bilatérales et asymétriques et siègent au niveau de la substance blanche périventriculaire postérieure, dans les radiations optiques, les centres semi-ovales et la substance blanche sous-corticale (fig. 22-11).
Ces atteintes constituent un large spectre clinique de déficits visuels pouvant être isolés ou associés. L’ophtalmologiste peut
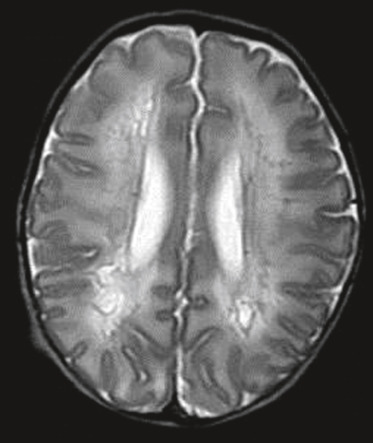
Fig. 22-11 Leucomalacie périventriculaire de forme kystique : coupe IRM séquence T2 axiale.
(Remerciements au Dr G. Soto Ares.)
être confronté à de nombreux cas de figure : chez le nouveauné à la phase aiguë ou subaiguë, ou plus tard chez l’enfant lourdement handicapé, les troubles ophtalmologiques sont sous-estimés, passant au second plan compte tenu de la gravité de l’état général ou de la difficulté à les distinguer les uns des autres. Chez l’enfant cérébrolésé moins lourdement handicapé, le comportement visuel peut paraître déroutant pour l’ophtalmologiste s’il n’en connaît pas les particularités. Enfin chez des enfants apparemment indemnes de toute séquelle neurologique, des troubles neurovisuels handicapants peuvent ne se révéler qu’à l’âge scolaire.
L’évaluation est importante pour guider la rééducation pluridisciplinaire (ophtalmologistes, neuropédiatres, neuropsychologues, radiologues, orthoptistes, etc.) et doit tenir impérativement compte de l’interrogatoire des personnes qui prennent soin de l’enfant au quotidien. Les troubles des enfants cérébrolésés sont en effet très fluctuants. Elle fait appel :
– à l’étude des réflexes psychovisuels (fixation, poursuite, attraction visuelle, préhension) avec des objets colorés, lumineux, sonores de taille et forme variables, dans des conditions et des ambiances différentes (éclairement, arrière-plan, etc.) ;
– à l’étude de la motilité automatique (réflexe vestibulo-oculaire et nystagmus optocinétique) ;
– aux techniques d’électrophysiologie visuelle (évaluation de l’acuité par les PEV par balayage ou PEV multivoies : voir chapitre 3.3).
Le comportement de cécité, principal motif de consultation chez le bébé, peut être dû, si la papille est normale, à une cécité corticale, une agnosie visuelle (absence d’intégration du message visuel), un retard de maturation visuelle ou une paralysie des mouvements conjugués des yeux (apraxie oculomotrice [AOM]). Si la papille est atrophique, il n’est pas simple de faire la part entre la déficience due à l’atteinte des voies visuelles antérieures et celle liée à l’atteinte des voies postérieures. C’est surtout l’évolution qui pourra permettre de trancher (voir chapitre 22.4). La préservation de la poursuite visuelle d’objet en verticalité fera le diagnostic différentiel en faveur d’une AOM. L’étude des réflexes pupillaires est en faveur d’une atteinte des voies visuelles antérieures s’ils sont absents (intérêt des PEV, voir chapitre 3.3).
Les troubles oculomoteurs sont les anomalies ophtalmologiques les plus fréquentes :
– chez les enfants nés à terme, l’atteinte corticale étant prédominante, les déviations horizontales conjuguées du regard et l’exotropie précoce sont fréquentes, le nystagmus est rare, intermittent, surtout constaté lors des mouvements conjugués des yeux ;
– chez les enfants prématurés, l’atteinte sous-corticale étant prédominante, les lésions sont responsables d’un(e) :
– déviation tonique du regard vertical, principalement observée dans les atteintes thalamiques et/ou mésencéphaliques et l’hydrocéphalie ;
– strabisme, le plus souvent convergent sauf dans les formes graves où il est généralement divergent, parfois divergent puis convergent (strabisme dyskinétique) ;
– nystagmus, plus fréquemment latent ;
– chez les enfants cérébrolésés, prématurés ou nés à terme, la prévalence du strabisme est très élevée (en moyenne 70 % ), la fixation peut être furtive, excentrée, instable ou absente lorsque la vision est périphérique donnant un aspect de strabisme divergent. Les anomalies de la motilité conjuguée sont quasi constantes : les poursuites sont saccadées, lentes, très lentes voire absentes, les saccades aussi bien réflexes que visuoguidées sont dysmétriques, mal calibrées ou absentes. Ces troubles sont souvent retrouvés lors des examens réalisés à l’âge scolaire soit parce qu’ils sont passés inaperçus dans la petite enfance, soit parce qu’ils ont été oubliés chez un enfant qui évoluait bien. Les paralysies oculomotrices sont plus rares, isolées ou multiples [4-12].
La malvoyance cérébrale et son aspect fluctuant relèvent de nombreuses causes décrites ci-dessous.
La baisse d’acuité visuelle est secondaire à l’atteinte des nerfs optiques (l’atteinte des voies visuelles est responsable de déficit du champ visuel et non de l’acuité et l’atteinte corticale est responsable d’une cécité). Elle peut aussi être due aux anomalies de la réfraction. Elle peut être évaluée par les PEV par balayage quand aucune autre méthode n’est possible.
Il s’agit :
d’amputations du CV qui sont faciles à détecter par les méthodes d’étude du CV par attraction :
hémianopsie latérale homonyme et quadranopsie latérale homonyme (souvent associées à une hémiplégie) des lésions rétrochiasmatiques ;
rétrécissement tubulaire séquellaire de la cécité corticale ou de l’atrophie optique (post-hydrocéphalie surtout) ;
déficit du CV inférieur par atteinte des radiations optiques supérieures : c’est le déficit le plus fréquent dans la LMPV, cause de chutes, à la descente d’escaliers notamment, et de problèmes à l’apprentissage de la lecture. Il faut garder à l’esprit que quand l’enfant ne dirige pas son regard vers une cible visuelle qui apparaît dans son CV, il peut s’agir d’un déficit du CV mais aussi d’un déficit du regard conjugué (apraxie oculomotrice surtout) ou d’une héminégligence.
de déficits du champ localisés au champ central ou mal systématisés avec des zones de vision et non-vision (déficits en gruyère) qui sont cause de maladresse, d’un comportement de malvoyance fluctuant avec l’attention et la fatigue, et de difficultés des apprentissages scolaires (lecture, calcul). L’analyse fine du comportement visuel, les tests neuropsychologiques adaptés à l’âge et à l’enfant et parfois la mise en évidence d’asymétrie aux PEV multivoies permettront d’évoquer le diagnostic quand le CV instrumental n’est pas réalisable.
Ces anomalies du CV peuvent s’améliorer avec l’âge soit du fait de la plasticité cérébrale, soit de l’amélioration de l’attention [4, 6, 11].
Les fluctuations de la vision chez les enfants cérébrolésés peuvent être dues à de nombreuses causes, soit communes à tous les enfants mais avec un plus fort retentissement (troubles d’attention aggravés par la fatigue, les maladies intercurrentes, intolérance au stress et aux bruits), soit spécifiques. Ces enfants, prématurés ou non, sont en général très photophobes, ont des difficultés à écouter et regarder en même temps, à distinguer les objets sur des fonds complexes, à percevoir les profondeurs (ils ont des difficultés à apprécier les différences entre un sol plat et des marches, et ils tâtonnent du pied lorsqu’ils passent d’un sol coloré à un autre) et à retrouver leur chemin. Ils voient parfois mieux les objets en mouvements (voies magnocellulaires) que les objets fixes, et mieux dans la pénombre qu’à la lumière ; ils identifient aussi parfois mieux la couleur des objets que leur forme. Ces troubles visuospatiaux sont souvent méconnus lorsqu’ils sont isolés et ne sont décelés que lors des premières exigences scolaires. Ils sont à l’origine des troubles de l’apprentissage d’où l’intérêt d’un diagnostic précoce. L’apraxie visuospatiale serait le trouble cognitif le plus fréquent chez l’enfant prématuré porteur de LMPV (voir chapitre 30.7) [13, 14].
La papille est normale ou modérément atrophique chez les nouveau- nés à terme. Dans la LMPV, la papille est souvent hypoplasique ou normale à grande excavation, très évocatrice de la LMPV due à l’atteinte des radiations optiques avec dégénérescence transynaptique rétrograde des voies visuelles (fig. 22-12) [4–6, 11, 15]. Pour une description détaillée, voir chapitre 21.1.
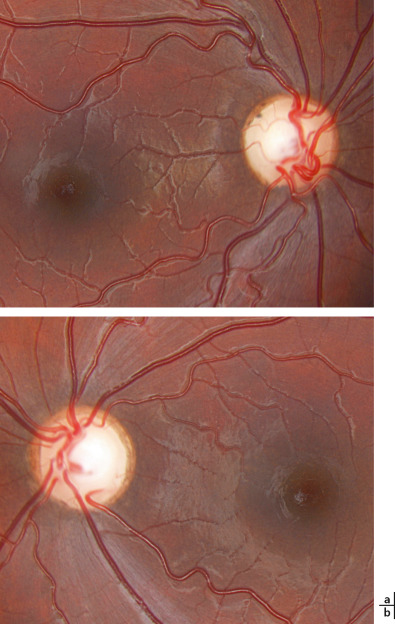
Fig. 22-12 a, b. Papilles de taille normale pâles et excavées chez un enfant prématuré né à 26 SA aux antécédents d’anoxie périnatale.
Ces anomalies sans rapport avec les lésions cérébrales vont être de traitement plus difficile chez les enfants cérébrolésés ou en compliquer le pronostic. Ce sont essentiellement la rétinopathie du prématuré et les anomalies de la réfraction dont la prévalence augmente avec le degré de prématurité. La myopie est la principale amétropie chez le prématuré, mais ce sont l’hypermétropie et l’astigmatisme qui sont plus souvent retrouvés chez l’ensemble des enfants qui ont une souffrance périnatale, ce qui impose leur dépistage précoce par la réalisation d’une réfraction sous cycloplégie. L’indication d’une correction optique ainsi que le traitement de l’amblyopie se font au cas par cas. La correction optique totale est indispensable chez l’enfant cérébrolésé qui a une amétropie à risque amblyogène. Chez les enfants lourdement handicapés, avec une amétropie faible ou moyenne, et chez les enfants qui n’ont qu’une vision périphérique, ou une déviation conjuguée du regard, elle sera interrompue si le comportement se dégrade depuis le port de la correction [6].
L’atteinte ophtalmologique est extrêmement fréquente dans la souffrance périnatale.
Le dépistage précoce des atteintes ophtalmologiques liées à la souffrance périnatale est nécessaire et doit être systématique. Il sera réalisé avant toute évaluation neuropsychologique des troubles neurovisuels. La prise en charge précoce pluridisciplinaire et adaptée permettra à ces enfants une intégration sociale et scolaire.
[1] Luca A, Ramenghi L, Bassi L, et al. Perinatal brain damage from pathogenesis to neuroprotection. Mariani Foundation Pediatric Neurology. John Libre Eurotext ; 2008, p. 55-63.
[2] Chalard F, Garel C, Ducou Le Pointe H. Imagerie de l’ischémie périnatale. MT Pédiatrie 2013 ; 16 : 203-11.
[3] Anthonioz C, Loisel D, Delorme B, et al. Aspects IRM de l’encéphalopathie anoxoischémique du nouveau-né à terme et du prémature. Journal de Radiologie 2006 ; 87 (11-c1).
[4] Brodsky MC. Pediatric neuroophthalmology. New York : Springer ; 2010
[5] Mosin IM, Moshetova LK, Slavinskaia NV, et al. Ophthalmologic symptomatology in children with periventricular leucomalation. Vestn Oftalmol 2005 ; 121 : 13-8.
[6] Jacobson LK, Dutton GN. Periventricular leukomalacia : an important cause of visual and ocular motility dysfunction in children. Surv Ophthalmol 2000 ; 45 : 1-13.
[7] Fazzi E, Signorini SG, La Piana R, et al. Neuro-ophthalmological disorders in cerebral palsy : ophthalmological, oculomotor, and visual aspects. Dev Med Child Neurol 2012 ; 54 : 730-6.
[8] Ozturk AT, Berk AT, Yaman A. Ocular disorders in children with spastic subtype of cerebral palsy. Int J Ophtalmol 2013 ; 6 : 204-10.
[9] Cioni G, Fazzi B, Coluccini M, et al. Cerebral visual impairment in preterm infants with periventricular leukomalacia. Pediatric Neurol 1997 ; 17 : 331-8.
[10] Salati R, Borgatti R, Giammari G, Jacobson L. Oculomotor dysfunction in cerebral visual impairment following perinatal hypoxia. Dev Med Child Neurol 2002 ; 44 : 542-50.
[11] Glass HC, Fujimoto S, Ceppi-Cozzio C, et al. White-matter injury is associated with impaired gaze in premature infants. Pediatr Neurol 2008 ; 38 : 10-5.
[12] Jacobson L, Ygge J, Flodmark O. Nystagmus in periventricular leucomalacia. Br J Ophthalmol 1998 ; 82 : 1026-32.
[13] Chokron S. Approche neuropsychologique des troubles neurovisuels chez l’enfant. Rev Neuropsychol 2015 ; 7 : 41-9.
[14] Dalens H. Les pathologies neurovisuelles chez les enfants cérébrolésés. Motricité Cérébrale 2014 ; 35 : 25-40.
[15] Brodsky MC. Semiology of periventricular leucomalacia and its optic disc morphology. Br J Ophthalmol 2003 ; 87 : 1309-10.
C. Marks
Chez un nourrisson adressé pour comportement de cécité, avec des réflexes photomoteurs normaux et un fond d’oeil normal ou n’expliquant pas la cécité, le diagnostic se pose entre cécité corticale, qui nécessite un bilan neuropédiatrique complet, et retard de maturation visuelle. Les pronostics visuels et généraux sont tout à fait différents et les PEV vont permettre de nous orienter vers l’une ou l’autre pathologie.
Un antécédent de souffrance périnatale est le facteur de risque le plus important de développer une cécité corticale chez un enfant [1]. Étant donné que le taux de survie chez les enfants nés prématurément est en augmentation, cette pathologie devient une cause importante de basse vision chez l’enfant. En effet, une leucomalacie périventriculaire (LMPV) affectant les radiations optiques est devenue une des principales causes de malvoyance cérébrale chez les enfants nés prématurément [2]. L’interrogatoire recherche des antécédents de souffrance périnatale ayant pu entraîner des lésions cérébrales : infection, ischémie, hypoxémie, hypoglycémie, désordres métaboliques, convulsions, etc. Cette atteinte des voies visuelles rétrochiasmatiques, au niveau occipital, entraîne un défaut de traitement de l’information visuelle à l’étage cognitif. Elle peut être associée, ou non, à une atteinte des voies visuelles antérieures responsable d’une atrophie optique.
Une absence de suivi oculaire horizontal et vertical chez un nourrisson qui semble indifférent aux stimulations visuelles, mais qui reste souriant et détendu, est en faveur d’un retard de maturation visuelle (vidéo 22.1).
Vidéo 22.1 - Retard de maturation visuelle: Nourrisson de 4 mois adressé pour absence de suivi oculaire. Le bilan électrophysiologique est en faveur d’un retard de maturation visuelle. Le comportement visuel se normalise à l’âge de 6 mois.
Si la poursuite est obtenue uniquement sur une mire qui se déplace verticalement, il faut évoquer une apraxie oculomotrice (vidéo 22.2), qui est un diagnostic différentiel d’une absence de suivi oculaire chez le nourrisson.
Vidéo 22.2 - Apraxie oculomotrice: Enfant de 18 mois adressée pour anomalie du comportement visuel avec absence de poursuites horizontales. L’examen oculomoteur fait poser le diagnostic d’apraxie oculomotrice.
L’existence d’une « errance du regard » , constituée de mouvements oculaires lents et pendulaires (vidéo 22.3), est toujours un signe de gravité et ne se retrouve que chez les nourrissons atteints d’une dystrophie rétinienne congénitale précoce, notamment une amaurose congénitale de Leber, ou d’une affection neurologique grave. Dans ces deux cas et contrairement à la cécité corticale ou au retard de maturation, les réflexes photomoteurs sont ralentis. De tels mouvements oculaires nécessitent de réaliser un ERG à l’âge de 4 mois.
Vidéo 22.3 - Errance du regard: Enfant de 2 mois. Amaurose congénitale de Leber.
L’examen en LAF recherche, dans le cadre du diagnostic différentiel, une transillumination irienne en faveur d’un albinisme,qui peut se manifester par un mauvais comportement visuel au cours des premières semaines de vie, avant que n’apparaisse un nystagmus [3].
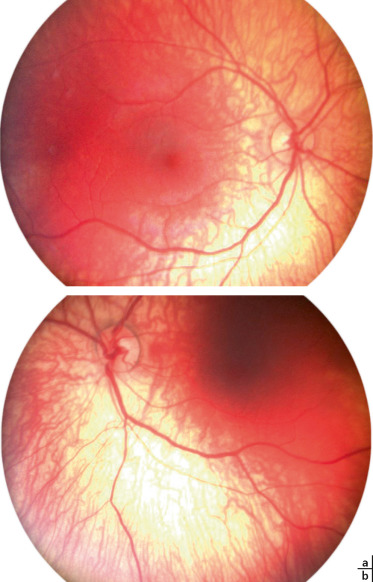
Fig. 22-13 Nourrisson de 3 mois adressé par son médecin traitant pour absence de suivi oculaire (a. oeil droit ; b. oeil gauche).
L’examen ophtalmologique ne retrouve pas de suivi sur une mire contrastée, les réflexes photomoteurs sont normaux, le FO montre une légère pâleur papillaire avec rétine globalement hypopigmentée, n’expliquant pas le comportement de cécité.
Le FO est normal ou montre une papille pâle, d’aspect « grisâtre » , en cas de retard de maturation (fig. 22-13) [4]. Chez les enfants polyhandicapés avec souffrance cérébrale diffuse, une atrophie optique peut être associée à la malvoyance cérébrale, ce qui rend difficile l’évaluation de la part de baisse d’acuité visuelle due à la cécité corticale, de celle due à l’atrophie optique.
– les PEV flashes peuvent être présents dans la cécité corticale et dans le retard de maturation, ils ont peu de valeur diagnostique. En l’absence d’atrophie optique, des PEV flashes plats, déstructurés, sont en faveur d’une cécité corticale (fig. 22-14). Parfois, le PEV ne peut être extrait de l’électroencéphalogramme, donnant un aspect déstructuré avec de nombreux rejets, qui persiste même lorsque la source lumineuse est détournée du regard de l’enfant, ce qui est un signe d’épilepsie associée (voir chapitre 3.3) ;
– les PEV par damiers ne sont interprétables de façon fiable que s’ils sont présents et sont alors un argument en faveur d’un retard de maturation et de bon pronostic (fig. 22-15) ; ils peuvent être absents si l’enfant ne regarde pas l’écran et doivent être recontrôlés ;
– une asymétrie croisée aux PEV flashes oriente vers un albinisme.
Au terme de cet examen clinique et électrophysiologique, l’ophtalmologiste devra décider de la réalisation, ou non, d’examens complémentaires comme une IRM cérébrale qui nécessite souvent une sédation après l’âge de 3 mois, et renseigner les parents sur le pronostic visuel de leur enfant.
Les facteurs en faveur d’un retard de maturation visuelle isolé sont : l’absence d’antécédent périnatal et/ou d’errance du regard, et la présence de PEV par damiers. Un examen clinique neuropédiatrique permettra de s’assurer que le développement psychomoteur est normal pour l’âge [7]. Des conseils de stimulation visuelle par des objets lumineux et des mires contrastées vont être donnés aux parents et une surveillance ophtalmologique est mise en place. Le comportement visuel commence à s’améliorer entre 3 et 4 mois et se normalise avant l’âge de 6 mois.
Une cécité corticale est évoquée chez un nourrisson avec des antécédents périnataux et des PEV flashes déstructurés. Un examen neuropédiatrique doit être réalisé et retrouve souvent un retard psychomoteur. Une IRM cérébrale doit également être demandée à la recherche de lésions occipitales (fig. 22-16) [8]. La récupération visuelle est très progressive et dépend de l’affection cérébrale à l’origine de la cécité corticale et de son traitement. L’enfant est d’abord très fortement attiré par la lumière, puis fixe de façon éphémère les visages et les objets en mouvement. Ces épisodes de fixation seront de plus en plus fréquents et prolongés au cours de la journée. Lorsque la souffrance céré-
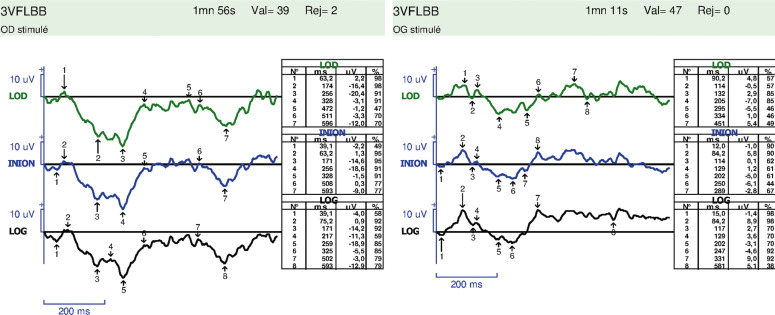
Fig. 22-14 Nourrisson de 20 mois hospitalisé pour inhalation de corps étranger à l’origine de six épisodes brefs d’arrêt cardiorespiratoire.
L’enfant est adressée pour comportement de cécité depuis le réveil du coma. Elle ne suit ni les visages, ni une mire contrastée ; les réflexes photomoteurs et le FO sont normaux. Les réponses aux PEV flashs sont déstructurées, en faveur d’une cécité corticale.
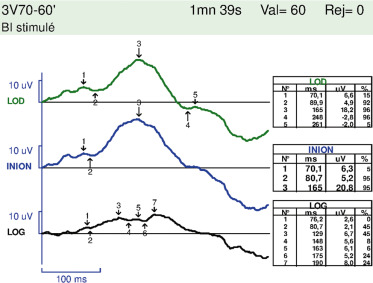
Fig. 22-15 Analyse des PEV chez le nourrisson présenté à la figure 22-13. Présence d’une réponse aux PEV 60´ est en faveur d’un retard de maturation visuelle.
Des conseils de stimulation visuelle sont donnés. Lors de la consultation de contrôle 3 mois plus tard, les parents décrivent une amélioration du comportement visuel, l’enfant sourit en réponse et attrape ses jouets. Les réponses au test du regard préférentiel sont symétriques entre les deux yeux et normales pour l’âge.
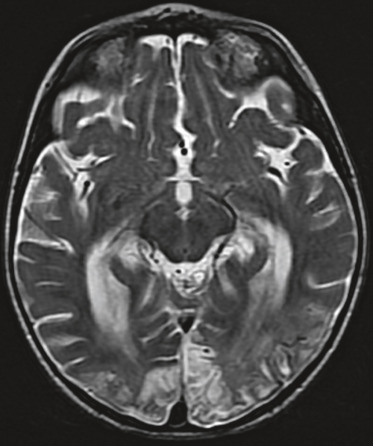
Fig. 22-16 IRM cérébrale (même enfant qu’à la figure 22-14) retrouvant des lésions diffuses bilatérales des régions occipitales et pariétales, et comportant des zones de nécrose corticale essentiellement au niveau des régions occipitales à prédominance gauche.
L’évolution est marquée par une bonne récupération visuelle, 6/10 aux deux yeux à 4 ans, avec persistance de troubles visuospatiaux.
brale n’a été que transitoire comme en cas d’hypoxémie acquise, le comportement visuel peut se normaliser plus rapidement mais il peut persister des troubles du champ visuel [9]. C’est à l’âge scolaire que peuvent survenir des difficultés liées à des troubles visuospatiaux séquellaires.
Enfin, en l’absence d’antécédent périnatal, une cécité corticale chez un nourrisson peut être d’origine infectieuse (méningo-encéphalite herpétique), traumatique (syndrome du bébé secoué), tumorale, secondaire à une hydrocéphalie ou à une épilepsie occipitale (syndrome de West) [8]. En cas de cécité corticale acquise chez un enfant qui présente des troubles du comportement et une régression psychomotrice, il faut rechercher une adrénoleucodystrophie liée à l’X, maladie neurologique rare secondaire à une démyélinisation de la substance blanche touchant principalement les aires pariéto-occipitales, les radiations optiques et le splénium du corps calleux [10].
Un antécédent de souffrance périnatale est le facteur de risque le plus important de développer une cécité corticale chez un enfant.
Une « errance du regard » est toujours un signe de malvoyance et ne se retrouve qu’en cas de dystrophie rétinienne précoce ou de cécité corticale avec affection neurologique grave.
Des PEV flashes normaux n’ont que peu de valeur diagnostique.
Des PEV flashes plats déstructurés orientent vers une cécité corticale (si FO normal).
Des PEV damiers sont de bon pronostic, en faveur d’un retard de maturation des voies visuelles.
Le FO peut être normal dans les deux cas, une atrophie optique peut être associée à la malvoyance cérébrale, une papille d’aspect « grisâtre » est décrite dans les retards de maturation.
[1] Van Genderen M, Dekker M, Pilon F, Bals I. Diagnosing cerebral visual impairment in children with good visual acuity. Strabismus 2012 ; 20 : 78-83.
[2] Jacobson LK, Dutton GN. Periventricular leukomalacia : an important cause of visual and ocular motility dysfunction in children. Surv Ophthalmol 2000 ; 45 : 1-13. Review.
[3] Tresidder J, Fielder AR, Nicholson J. Delayed visual maturation : ophthalmic and neurodevelopmental aspects. Dev Med Child Neurol 1990 ; 32 : 872-81.
[4] Hoyt CS, Jastrzebski G, Marg E. Delayed visual maturation in infancy. Br J Ophthalmol 1983 ; 67 : 127-30.
[5] Defoort S. Potentiels évoqués visuels de l’enfant. In : Riss JF. Exploration de la fonction visuelle. Rapport de la Société Française d’Ophtalmologie 1999, p. 629-37.
[6] Creel DJ. Visually evoked potentials. In: Kolb H, Fernandez E, Nelson R. Webvision : The Organization of the Retina and Visual System. Salt Lake City (UT) : University of Utah Health Sciences Center ; 1995.
[7] Azmeh R, Lueder GT. Delayed visual maturation in otherwise normal infants. Graefes Arch Clin Exp Ophthalmol 2013 ; 251 : 941-4.
[8] Philip SS, Dutton GN. Identifying and characterising cerebral visual impairment in children : a review. Clin Exp Optom 2014 ; 97 : 196-208.
[9] Guzzetta A, D’Acunto G, Rose S, et al. Plasticity of the visual system after early brain damage. Dev Med Child Neurol 2010 ; 52 : 891-900. Review.
[10] Zgorzalewicz-Stachowiak M, Stradomska TJ, Bartkowiak Z, Galas-Zgorzalewicz B. Cerebral childhood and adolescent X-linked adrenoleukodystrophy. Clinical presentation, neurophysiological, neuroimaging and biochemical investigations. Folia Neuropathol 2006 ; 44 : 319-26.
I. Drumare-Bouvet
Nous nous intéresserons essentiellement aux tumeurs qui affectent les voies optiques après le canal optique (gliomes des voies optiques et craniopharyngiomes), et nous citerons les tumeurs de la fosse postérieure et du tronc cérébral qui donnent essentiellement des troubles oculomoteurs, et les autres tumeurs cérébrales qui entraînent une hypertension intracrânienne (HTIC) et ou des troubles du champ visuel (CV).
Les tumeurs cérébrales sont les tumeurs solides les plus fréquentes de l’enfant, et elles arrivent en deuxième position après les leucémies. Tous cas confondus, elles concernent 30 cas par million d’enfants. On distingue deux pics de fréquence : chez le nourrisson, puis chez l’enfant plus grand entre 10 et 14 ans [1].
Le diagnostic initial de tumeur cérébrale est souvent difficile et le retard moyen au diagnostic est de quelques mois (20 semaines) [2]. Les signes cliniques varient en fonction du type de tumeur et de sa localisation. Ceux-ci sont souvent peu spécifiques.
Toutes sortes de céphalées peuvent révéler une tumeur cérébrale. On retiendra essentiellement leur aggravation progressive en intensité et en fréquence, leur horaire en fin de nuit, réveillant le malade le matin, et leur caractère rebelle aux antalgiques. Elles sont présentes dans 33 % des cas [3].
Les vomissements en jet, le matin, soulageant la céphalée, sont très caractéristiques, mais peuvent accréditer à tort une suspicion de troubles psychosomatiques.
Chez le petit enfant, leur intensité peut entraîner un comportement prostré, d’enfant excessivement sage qui n’ose plus bouger par peur des douleurs.
Chez le nourrisson, l’HTIC entraîne une disjonction des sutures et une augmentation du périmètre crânien [4].
Le FO confirme le diagnostic en objectivant l’oedème papillaire de stase : les vaisseaux sont noyés dans l’oedème, il peut exister des hémorragies péripapillaires et des plis rétiniens secondaires à l’oedème, voire un exsudat maculaire si l’oedème est ancien et important (fig. 22-17).
Les signes cliniques varient en fonction de la localisation de la lésion : des troubles de l’équilibre et de la coordination, un torticolis ou une ataxie évoquent une localisation en fosse postérieure (cervelet en particulier ; fig. 22-18) ; une paralysie des nerfs crâniens évoque une atteinte du tronc cérébral ; des troubles endocriniens orientent vers une tumeur de la ligne médiane ; et surtout un nystagmus, un strabisme, une baisse de vision voire des troubles du CV orientent vers un gliome des voies optiques .
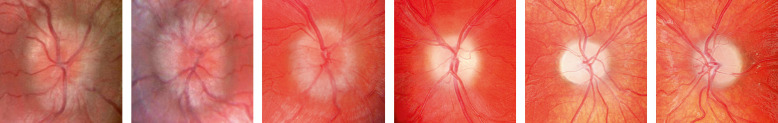
Fig. 22-17 Modifications papillaires d’origine tumorale.
a. Enfant de 10 ans, gliome infiltrant de la lame tectale, oedème papillaire bilatéral. b. Enfant de 6 ans, tumeur cérébrale avec HTIC, en postopératoire : oeil droit (OD), oedème papillaire persistant ; oeil gauche (OG), papille atrophique à bords flous. c. Enfant de 4 ans, astrocytome pilocytique grade I du cervelet, retard au diagnostic : atrophie optique post-stase et cécité légale au moment du diagnostic.
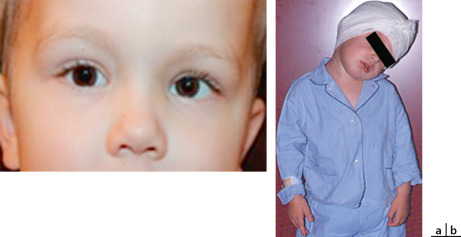
Fig. 22-18 Signes cliniques révélateurs de tumeur cérébrale.
a. Strabisme. b. Torticolis révélateur d’un astrocytome pilocytique du cervelet.
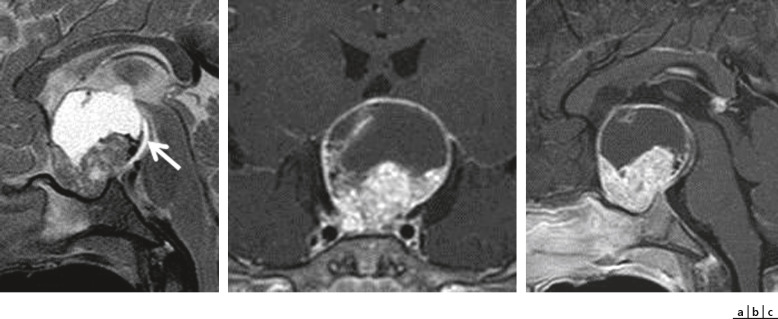
Fig. 22-19 a-c. IRM : craniopharyngiome chez un enfant de 9 mois qui avait un nystagmus horizontal de faible amplitude mais un bon comportement visuel.
Calcifications (flèche) et lésion volumineuse à l’IRM.
En urgence, un scanner cérébral permet le diagnostic dans la plupart des cas et oriente la prise en charge immédiate dans un centre spécialisé. En outre, il permet de voir des calcifications (évocatrices de craniopharyngiome). Dans un second temps, ou en urgence si cela est possible, l’IRM fait le diagnostic, précise la taille de la lésion, sa topographie, l’existence de lésions secondaires, d’une HTIC et donne de précieux renseignements sur le type histologique probable de la tumeur (fig. 22-19).
(À l’exclusion du gliome du nerf optique qui est traité dans le chapitre 21.5.)
Le gliome des voies optiques appartient au groupe hétérogène des gliomes de bas grade. C’est une tumeur rare qui représente 4 à 6 % des tumeurs cérébrales de l’enfant. Il survient dans 90 % des cas avant l’âge de 20 ans (c’est une tumeur de l’enfant), dont dans 75 % des cas avant l’âge de 10 ans, et dont dans 60 % des cas avant l’âge de 5 ans. Dans environ 50 % des cas, il est associé à la neurofibromatose de type 1 (NF1), mais seules 1 à 5 % des NF1 ont un gliome. Le taux de survie est supérieur à 90 % à 10 ans, mais dans 40 % des cas son évolution est émaillée de récidives. Le pronostic de ces tumeurs est donc essentiellement fonctionnel et en particulier grevé par les complications visuelles et endocriniennes (fig. 22-20) [1, 4-7].
Pour guider la prise en charge de ces tumeurs, une classification topographique établie par Dodge en 1958 [4] puis modifiée par les données de l’imagerie, reste la plus utilisée :
– type I : la tumeur est limitée à un seul nerf optique dans sa portion intra-orbitaire (voir chapitre 21.5) ;
– type II : la tumeur envahit en partie ou en totalité le chiasma, avec ou sans envahissement des nerfs optiques, mais sans envahissement postérieur ou supérieur ;
– type III : la tumeur infiltre le chiasma avec extension aux structures de voisinage (V3 ou hypothalamus, tractus optiques), avec ou sans atteinte des nerfs optiques
Ces tumeurs sont le plus souvent des astrocytomes pilocytiques, parfois des astrocytomes pilomyxoïdes monomorphes, qui se rencontrent plutôt chez le jeune enfant et sont plus agressifs. Actuellement, il est recommandé de biopsier ces tumeurs quand le patient n’a pas de NF1. À l’inverse, en cas de NF1 connue et si les images radiologiques sont typiques, on ne fera pas de biopsie.
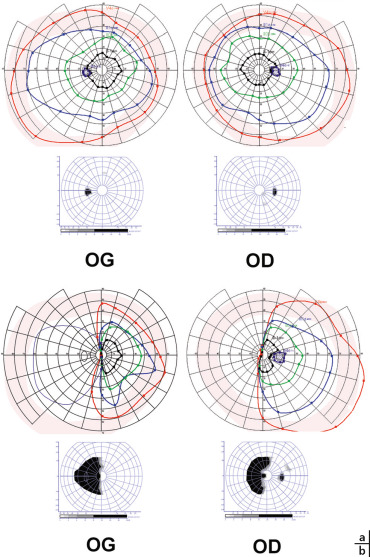
Fig. 22-20 Recul de 10 ans chez deux jeunes filles âgées de 15 ans atteintes de gliome du chiasma
a. Éloïse : statut NF1, 3 lignes de chimiothérapie, acuité visuelle (AV) 10/10 oeil droit et gauche (ODG), aucune séquelle campimétrique, pas d’invalidité. b. Jeanne : pas de NF1, 2 lignes de chimiothérapie et chirurgie, AV 10/10 ODG, hémianopsie latérale homonyme gauche qui contre-indique la conduite et complique l’autonomie.
Les circonstances de découverte sont essentiellement visuelles, sauf en cas d’IRM systématique dans le cadre de la surveillance d’une NF1.
– Nystagmus chez un nourrisson : il est présent dans 23 % des cas au moment du diagnostic [5]. Il peut être de tout type : horizontal, vertical, rotatoire, à bascule, mono- ou binoculaire. Il peut prendre l’aspect d’un spasmus nutans, c’est-à-dire d’un nystagmus bilatéral le plus souvent, rapide, de faible amplitude (en « aile d’abeille » ), associé à un nystagmus de la tête et à un torticolis. Il est plus particulièrement évocateur s’il est vertical, monoculaire ou associé à des signes digestifs, conduisant à un amaigrissement important avec cassure de la courbe pondérale, qui réalisent la cachexie diencéphalique de Russel (fig. 22-21). Ce tableau doit être bien connu car les troubles digestifs étant souvent sévères, ils masquent le diagnostic, or il correspond à des gliomes sévères pouvant mettre en jeu le pronostic vital (gliome avec atteinte du plancher du V3).
– Baisse d’acuité visuelle : une atteinte de la fonction visuelle serait présente dans 88 % des cas au moment du diagnostic, uniou bilatérale (fig. 22-22) [5. Néanmoins elle est difficile à mettre en évidence chez le très jeune enfant et n’est donc pas dans ce cas une circonstance de découverte.
– Strabisme, surtout s’il est acquis, convergent ou divergent.
– Au cours de la surveillance d’une NF1 (voir chapitre 20.10) : la fréquence des gliomes dans la NF1 justifie une surveillance régulière et surtout très précoce chez les patients atteints puisque la fréquence de ces tumeurs qui est maximale avant 2 ans décroît avec l’âge et devient très faible après 10 ans.
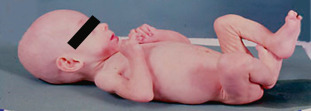
Fig. 22-21 Syndrome de Russel.
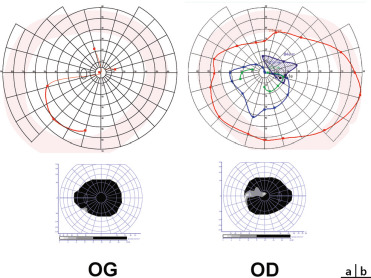
Fig. 22-22 Jeune fille âgée de 15 ans adressée pour baisse d’acuité visuelle de l’OG : OD 10/10 ; OG perception lumineuse..
a. CV agonique ODG ; FO : pâleur papillaire. b. Perte en fibres sévère à l’OCT, révélateur d’un volumineux gliome du chiasma optique (non-NF1).
– Signes endocriniens : ils sont rarement des circonstances de découverte du gliome des voies optiques, mais sont présents dans 26 % des cas au moment du diagnostic [7]. L’atteinte la plus fréquemment présente au moment du diagnostic est la puberté précoce suivie par le déficit en hormone de croissance. On peut également trouver une obésité, un diabète insipide ou une insuffisance anté-hypophysaire.
– Signes neurologiques : essentiellement l’HTIC, les troubles de l’apprentissage ou du comportement et le retard psychomoteur.
– Le FO peut être normal à tous les stades de la maladie et être donc faussement rassurant, notamment dans le cadre de la surveillance d’une NF1(fig. 22-23).
– L’oedème papillaire, uni- ou bilatéral selon la topographie de la lésion, peut aussi traduire une HTIC (fig. 22-24).
– L’atrophie optique peut également être uni- ou bilatérale, sectorielle ou totale, primaire (survenant sans oedème papillaire préalable) ou secondaire à un oedème papillaire.
– IRM : c’est l’examen de choix pour le diagnostic des gliomes des voies optiques, et pour le suivi et l’évaluation du traitement. Son aspect est celui d’une masse nodulaire ou infiltrante avec des prises de contraste homogènes et des kystes fréquents (fig. 22-25).
– Champ visuel (CV) : l’incidence d’atteinte du CV est sans doute au moins de 63 % [6], mais est en fait difficile à évaluer avec précision compte tenu du jeune âge de ces enfants et de la fréquence des troubles de l’attention et de la concentration quand il y a une NF1 [8]. Il peut être réalisé au moyen de procédures automatisées ou manuelles (Goldman). Il faut étudier non seulement le CV central mais aussi le CV périphérique ; chez le grand enfant, il a un intérêt majeur pour le suivi de l’évolution de la pathologie et pour l’évaluation des aptitudes professionnelles, à la conduite automobile ou du handicap.
– PEV [5] : leur place reste controversée tant pour le diagnostic que pour le suivi des gliomes des voies optiques, sans doute parce qu’ils peuvent être longs et délicats à réaliser chez le petit et nécessitent une équipe spécialisée tant pour la réalisation que pour l’interprétation. Néanmoins l’absence de sédation et la reproductibilité leur donnent un intérêt important dans cette pathologie. En effet, ils sont rapidement altérés en cas de souffrance des voies optiques mais restent normaux si la lésion ne déstructure pas les voies optiques. Leur altération dès la stimulation par flashes attire l’attention vers une atteinte des voies optiques chez un enfant qui a une amblyopie ou un nystagmus. Ils permettent de suivre régulièrement les jeunes enfants atteints de NF1 et d’éviter une IRM tant qu’ils sont normaux. Leur variation chez un patient traité (dégradationou amélioration) joue un rôle dans la décision thérapeutique, plus que l’évaluation de l’acuité visuelle dont les variations ne suivent pas toujours les variations de volume tumoral (fig. 22-26).
– Tomographie par cohérence optique (optical coherence tomography [OCT]) : pour certains [5, 9], l’OCT est rassurant quand l’épaisseur des fibres rétiniennes est normale, car elle est bien corrélée à la normalité de l’acuité visuelle et du CV. À l’inverse, une altération n’est pas synonyme de dysfonction visuelle, et seule une progression de plus de 10 % de la perte en fibres sur un ou plusieurs quadrants pourrait être le signe d’une aggravation de la pathologie. Néanmoins chez le petit enfant, l’OCT doit être réalisé avec un appareil portable et avec une sédation, ce qui limite son intérêt pour le suivi.
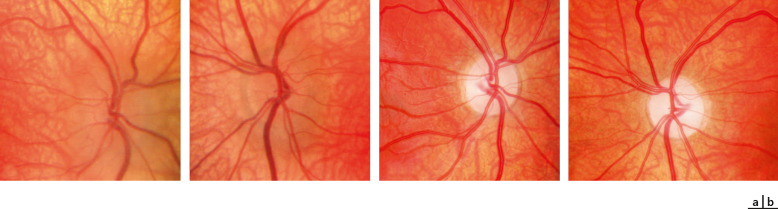
Fig. 22-23 Nourrisson de 5 mois, syndrome de Russel.
a. Au moment du diagnostic papilles subnormales. b. Après 5 ans d’évolution, malgré le traitement, atrophie optique bilatérale AV OD 1/20, OG 1/32.
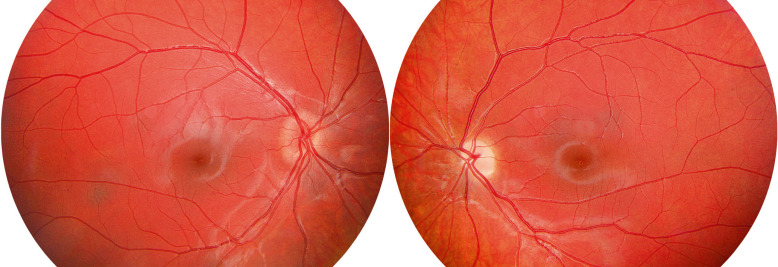
Fig. 22-24 Enfant de 5 ans, strabisme depuis 6 semaines et amblyopie gauche.
Découverte d’une NF1 familiale (l’enfant n’ayant aucun signe cutané) ; IRM ; gliome du chiasma étendu au nerf optique gauche ; traitement refusé par les parents ; après rééducation de l’amblyopie organique gauche récupération AV 10/10 ODG et CV normal.
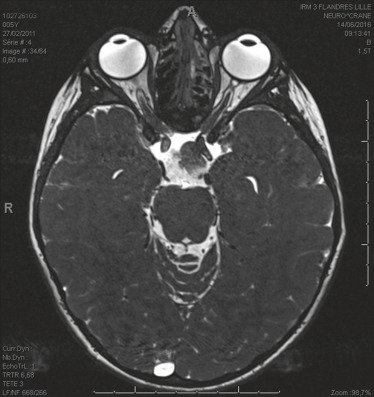
Fig. 22-25 Volumineux gliome des voies optiques du chiasma avec extension rétrochiasmatique, hypothalamique, bipallidale et protubérantielle droite chez une enfant de 5 ans NF1.
Une fois le diagnostic de gliome des voies optiques posé, il faut rechercher des signes de NF1, afin d’évaluer l’intérêt d’une biopsie pour poser un diagnostic histologique.
Les gliomes asymptomatiques seront simplement surveillés, car un certain nombre d’entre eux sont non évolutifs, voire peuvent involuer et même disparaître spontanément.
La chirurgie est indiquée pour traiter une HTIC ou à visée de réduction tumorale dans les formes très volumineuses ou kystiques.
La radiothérapie n’est en général plus proposée du fait des séquelles intellectuelles et endocriniennes qu’elle induit. Néanmoins elle peut être indiquée en cas de récidives.
La chimiothérapie est proposée en cas d’augmentation du volume tumoral sur deux IRM (fig. 22-27) successives ou en cas de signes fonctionnels ophtalmologiques. Elle a modifié le pronostic vital des formes III du petit enfant étendues au tractus visuel. Son efficacité sur la réduction du volume tumoral est reconnue, mais n’est pas démontrée en ce qui concerne l’amélioration de la fonction visuelle [8, 10].
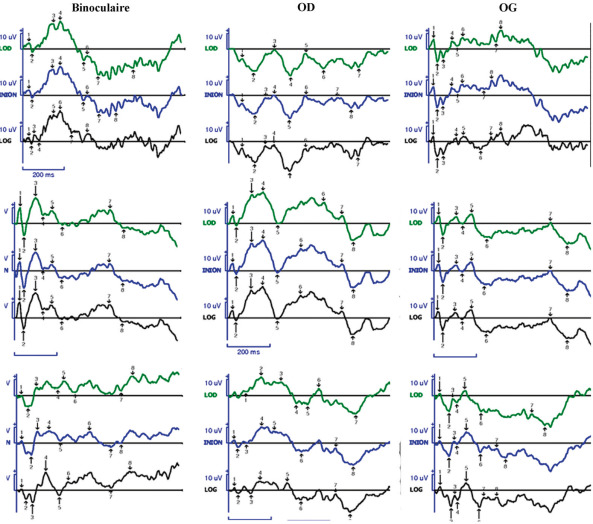
Fig. 22-26 Apport des PEV à la décision thérapeutique. Fillette âgée de 22 mois non-NF1, gliome des voies optiques découvert devant un nystagmus monoculaire droit.
a. PEV déstructurés ; traitement chimiothérapie. b. > 3 ans : arrêt du traitement ; on note une amélioration du tracé des PEV. c. 4 ans, augmentation du contraste à l’IRM, acuité stable, dégradation des PEV (enfant compliante), donc reprise du traitement.
Le craniopharyngiome est une tumeur épithéliale bénigne, prenant naissance au niveau de la tige pituitaire de l’hypophyse [7], développée dans les régions sellaire et suprasellaire. Il est une des tumeurs suprasellaires les plus fréquentes après le gliome des voies optiques, avec une prévalence de 1/200 000 [4]. Il représente 8 à 13 % des tumeurs de l’enfant (dont 54 % des tumeurs sellaires et chiasmatiques), avec un pic de fréquence entre 7 et 13 ans [11].
Dans 75 % des cas, ce sont les signes ophtalmologiques qui révèlent la tumeur :
– chez l’enfant, dans 30 % des cas ce sera une amblyopie bilatérale (50 % ), ou unilatérale, qui, fait majeur, ne s’améliore pas malgré un traitement bien conduit ;
– chez l’adolescent, on constate plutôt une baisse d’acuité visuelle ou des anomalies du CV ;
– un strabisme récemment acquis ;
– exceptionnellement un nystagmus chez le nourrisson.
Les signes endocriniens sont présents dans 63 % des cas au moment du diagnostic mais sont souvent sous-estimés : retard de croissance, obésité, retard pubertaire, diabète insipide
Enfin, parfois des signes neurologiques peuvent alerter, en particulier des troubles de mémoire ou une bradypsychie, voire des crises d’épilepsie. Une HTIC est très fréquemment présente.
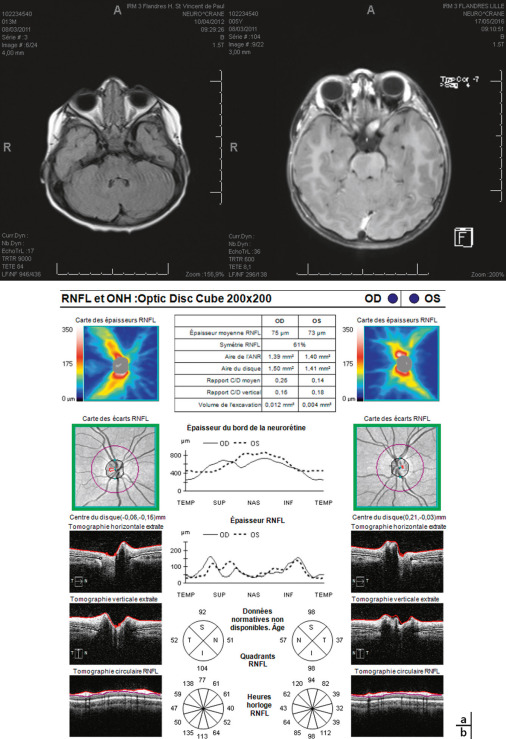
Fig. 22-27 Fille, 5 ans atteinte de NF1 : forte augmentation de volume du gliome à deux IRM à 3 ans d’écart (a) ; pas de retentissement visuel ; OCT normal (b).
Le FO peut paraître normal, même en cas de gliome volumineux au stade de début ou retrouver une pâleur papillaire ou une atrophie optique uni- ou bilatérale, sectorielle ou totale.
– CV : il est l’examen le plus important, tant pour orienter le diagnostic que pour le suivi de ces enfants et l’évaluation des séquelles. Les déficits sont en rapport avec la topographie de la lésion (essentiellement hémianopsie bitemporale, mais parfois latérale homonyme si la compression par la tumeur est latéralisée) [12]. Il faut étudier le CV central et le CV périphérique, ainsi que le CV binoculaire pour établir le certificat maison départementale des personnes handicapées (MDPH) et évaluer les aptitudes professionnelles ou les déplacements (vélo, conduite ultérieurement ; fig. 22-28). Néanmoins, il est difficilement réalisable de façon fiable avant l’âge de 6 ans.
– OCT : il semble que l’analyse du complexe ganglionnaire maculaire soit plus sensible pour dépister un retentissement sur les voies otiques que le retinal nerve fiber layer (RNFL). L’OCT est réalisable dans de bonnes conditions chez l’enfant sans sédation dès l’âge de 3 ans, ce qui en fait un examen intéressant pour le suivi des petits jusqu’à l’âge du CV.
– IRM cérébrale : c’est l’examen qui fait le diagnostic, permet de poser l’indication thérapeutique et de dépister d’éventuelles récidives. On note habituellement trois composantes dans ces tumeurs : kystique, calcifiée et solide (fig. 22-19).
Le traitement est chirurgical et consiste en une exérèse radicale s’il n’y a pas d’envahissement hypothalamique. Le cas échéant, elle sera complétée par une radiothérapie conformationnelle
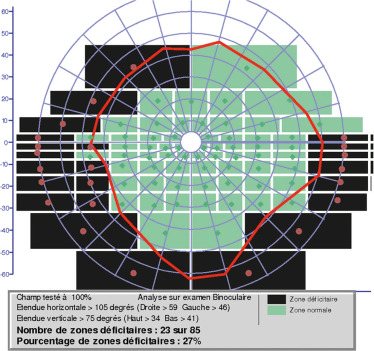
Fig. 22-28 Retentissement du craniopharyngiome chez un jeune homme de 25 ans (11 ans d’évolution).
AV OD 10/10, OG perception lumineuse, rendant la conduite impossible, associé au surpoids, à des troubles mnésiques et bradypsychie.
La survie à 5 ans est supérieure à 80 % , mais la morbidité de ces tumeurs est sévère [7, 12, 13]. Quatre-vingts pour cent des patients ont des complications visuelles (acuité visuelle et CV), essentiellement secondaires à la compression des voies optiques par le craniopharyngiome, alors que 84 % des patients ont des complications endocriniennes essentiellement secondaires au traitement. Les complications visuelles sont peu décrites chez l’enfant et certainement sous-estimées, mais elles semblent plus fréquentes que chez l’adulte.
L’association de la déficience visuelle, des troubles endocriniens, des troubles mnésiques, des anomalies du comportement et du caractère et de la baisse des performances scolaires génère chez ces patients une moins bonne qualité de vie que les autres malvoyants (fig. 22-28).
On citera les tumeurs hémisphériques (dysembryoplastic neuroepithelial tumour [DNET], primitive neuroectodermal tumours [PNET], desmoplastic infantile ganglioglioma [DIG], gliomes de haut et bas grade, etc.) qui sont responsables d’HTIC et de troubles du CV, et les tumeurs pinéales qui donnent également une hypertension intracrânienne et des troubles oculomoteurs à type de syndrome de Parinaud ou nystagmus retractorius.
Ces tumeurs sont révélées essentiellement par un torticolis, une raideur de nuque, une HTIC avec oedème papillaire qui peut générer une atrophie optique post-stase et des troubles oculomoteurs de type cérébelleux (nystagmus évoqué du regard, anomalies des saccades, apraxie oculomotrice). Il s’agit essentiellement d’astrocytomes pilocytiques du cervelet, de médulloblastomes, d’épendymomes ou de gliomes du tronc cérébral qui génèrent des paralysies oculomotrices fréquemment révélatrices (strabisme et diplopie).
Les tumeurs cérébrales après le chiasma optique sont les tumeurs solides les plus fréquentes de l’enfant. Elles sont néanmoins une pathologie rare, le plus souvent révélées par un tableau d’HTIC, qui est encore fréquemment mal identifié et conduit à un retard de diagnostic, ou par des signes ophtalmologiques au premier rang desquels on note le nystagmus précoce et la baisse d’acuité visuelle ou l’amblyopie réfractaire au traitement.
Par argument de fréquence, on retiendra :
le gliome des voies optiques, dont il faudra préciser le statut NF1 ou non-NF1, car les circonstances de découverte, la prise en charge et l’évolution diffèrent ;
le craniopharyngiome, tumeur bénigne qui entraîne des séquelles visuelles et endocriniennes sévères. Il est souvent diagnostiqué tard du fait de la discrétion des signes initiaux (amblyopie, retard de croissance) ;
On rappelle l’intérêt du CV pour la surveillance de ces tumeurs et en particulier le dépistage des récidives tumorales.
[1] Grill J, Roujeau T, Bouchireb K, et al. Tumeurs du système nerveux central Neurologie pédiatrique. Médecine Sciences-Flammarion ; 2010.
[2] Edgeworth J, Bullock P, Bailey A, et al. Why are brain tumors still being missed Arch Dis child 1996 ; 74 : 148-51.
[3] Wilne S, Collier J, Kennedy C, et al. Presentation of childhood CNS tumors : a systematic review and meta-analysis. Lancet Oncol 2007 ; 8 : 685-95.
[4] Dodge HW Jr, Love JG, Craig WM, et al. Gliomas of the optic nerves. AMA Arch Neurol Psychiatry 1958 ; 79 : 607-21.
[5] Dutton JJ. Gliomas of the anterior visual pathway. Surv Ophtalmol 1994 ; 38 :427- 52.
[6] Kelly JP, AH Weiss. Detection of tumor progression in optic pathway glioma with and without neurofibromatosis type 1. Neurooncology 2013 ; 15 : 1560-7.
[7] Robert M. Atteinte des voies visuelles chiasmatiques et rétrochiamatiques. Segment postérieur neuro-ophtalmologie. Médecine sciences-Flammarion/Lavoisier ; 2014
[8] Thoorens V. Pronostic visuel des gliomes chiasmatiques ; étude rétrospective de 59 cas [thèse doctorat de médecine]. Lille ; 2011.
[9] Avery RA, Cnaan A, Schuman JS, et al. Intra- and inter-visit reproducibility of ganglion cell-inner plexiform layer measurements using handheld optical coherence tomography in children with optic pathway gliomas. Am J Ophthalmol 2014 ; 158 : 916-23.
[10] Moreno L, Bautista F, Ashley S, et al. Does chemotherapy affect the visual outcome in children with optic pathway glioma. Sysematic review of the evidence. Eur J Cancer 2010 ; 46 : 2253-9.
[11]Hermann L. Muller Childhood craniopharyngioma. Pituitary 2013 ; 16 : 56-67.
[12] Defoort-Dhellemmes S, Moritz F, Bouacha I, Vinchon M.Craniopharyngioma : ophthalmological aspects at diagnosis. J Pediatr Endocrinol Metab 2006 ; 19 Suppl 1 : 321-4.
[13] Lo AC, Howard AF, Nichol A, et al. Long-term outcomes and complications in patients with craniopharyngioma : the British Columbia Cancer Agency experience. Int J Radiat Oncol Biol Phys 2014 ; 88 : 1011-8.
C. Marks
Les céphalées sont fréquentes chez l’enfant et l’adolescent. Un examen ophtalmologique est souvent demandé dans le cadre de leur bilan étiologique, dans la crainte d’une possible HTIC et pour éliminer une origine ophtalmologique.
Les migraines et les céphalées de tension sont les céphalées de l’enfant les plus fréquentes et représentent les céphalées primaires. L’International Headache Society (IHS) les différencie des céphalées secondaires symptomatiques. Il existe également des variantes de migraines chez l’enfant, encore appelées syndromes périodiques de l’enfance ou équivalents migraineux, et considérées comme des stades précurseurs de cette maladie. Leur diagnostic peut être difficile à un jeune âge.
La migraine est la cause la plus fréquente de céphalées chez l’enfant. Sa prévalence est d’environ 3 % à l’âge préscolaire et elle augmente avec l’âge, elle est estimée entre 4 et 11 % chez les enfants scolarisés à l’école primaire [1]. Pour poser le diagnostic de migraine, il est recommandé d’utiliser les critères de l’IHS. Chez l’adulte, l’interrogatoire doit retrouver la survenue d’au moins cinq crises de céphalées durant 4 à 72 heures, remplissant au moins deux des critères suivants : unilatérale, pulsatile, d’intensité modérée à sévère, s’aggravant à l’effort physique, et s’accompagnant soit de nausées, vomissements, soit de photophobie ou phonophobie [2]. Chez l’enfant, le seuil de durée des crises est plus court (1 heure), la localisation est frontale et bilatérale, les signes digestifs sont souvent au premier plan et une pâleur inaugurale est fréquemment retrouvée [3–5]. L’examen clinique, en particulier neurologique, doit être normal entre les crises.
Il s’agit d’une seconde forme de migraine qui s’accompagne de signes neurologiques focaux réversibles. L’aura est dans plus de 90 % des cas visuelle, se manifestant par des symptômes positifs (phosphènes) et/ou négatifs (perte de vision centrale ou flou visuel, hémianopsie latérale homonyme) totalement réversibles. Le scotome central scintillant, multicolore, s’étendant progressivement vers la périphérie du CV, affectant les deux yeux et souvent formé de lignes brisées, est caractéristique (fig. 22-29). L’aura migraineuse peut également se manifester par des symptômes sensitifs ou une aphasie. Ces signes neurologiques apparaissent progressivement en 5 minutes ou plus et durent jusqu’à 60 minutes. Ils peuvent être suivis d’une céphalée de type migraine, d’une céphalée de tension ou rester isolés (aura sans céphalée) [2].
La migraine rétinienne est une forme rare de migraine, survenant chez environ 1/200 patients migraineux. Elle se définit par des symptômes visuels monoculaires transitoires à type de phosphènes, scotome central, voire plus rarement une cécité. Ces signes peuvent être soit objectivés par un examen du CV pendant la crise, soit dessinés par le patient, et ne sont pas toujours associés à des céphalées [6]. Ils apparaissent progressivement en 5 minutes ou plus, et durent entre 5 et 60 minutes. S’ils sont accompagnés de céphalées, celles-ci surviennent dans les 60 minutes. Leur survenue nécessite un examen ophtalmologique complet, qui doit être normal entre les crises, excluant en particulier un processus embolique rétinien.
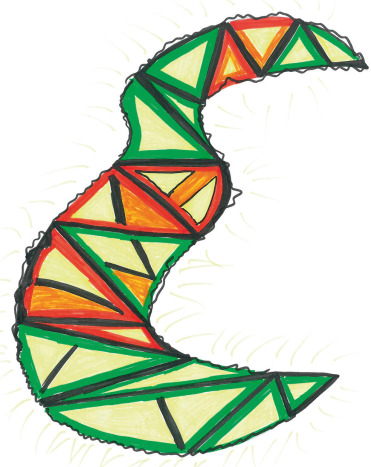
Fig. 22-29 Aura visuelle à type de scotome scintillant multicolore, dessiné par une adolescente de 14 ans.

Fig. 22-30 Modifications de la perception des visages précédant une céphalée migraineuse chez une adolescente.
Les équivalents migraineux ou syndromes périodiques de l’enfance correspondent à des déficits neurologiques périodiques et paroxystiques, parmi lesquels : le torticolis bénin paroxystique de l’enfance, le vertige bénin paroxystique de l’enfance, la migraine abdominale, les vomissements cycliques. L’interrogatoire retrouve souvent une histoire familiale de migraine chez un parent du premier ou du deuxième degré, et l’examen clinique neurologique est normal entre les crises [7, 8]. Ces équivalents migraineux sont considérés par l’IHS comme des stades précurseurs de migraines. La migraine confusionnelle aiguë et le syndrome d’ « Alice au pays des merveilles » sont des formes rares qui ne sont pas reconnues par l’IHS [5]. Le syndrome d’Alice au pays des merveilles est considéré comme une aura migraineuse et est souvent suivi d’une céphalée. Il a été décrit pour la première fois en 1952 par Lipman dont un patient se sentait « petit et élargi » , en référence aux frères Tweedle représentés dans le conte Alice au pays des merveilles. Il semble que Lewis Caroll souffrait lui-même de migraines. Les enfants atteints de ce syndrome souffrent d’épisodes d’hallucinations visuelles se manifestant par des distorsions en taille ou en forme des objets (micropsies, macropsies, métamorphopsies), des modifications de l’image corporelle et de la perception de l’environnement (fig. 22-30). Ils ne sont pas effrayés par leurs symptômes, ne présentent pas d’altération de la conscience, et retrouvent leur état antérieur après la crise [9].
Les céphalées de tension se distinguent des migraines par leur caractère plus diffus, bilatéral et non pulsatile, à type de pression.Leur intensité est légère à modérée et n’est pas aggravée par les efforts physiques. La survenue d’au moins 10 épisodes ayant duré de 30 minutes à 7 jours permet de poser le diagnostic. Le diagnostic différentiel entre migraines et céphalées tensionnelles peut être difficile chez l’enfant puisque les migraines sont responsables de céphalées bilatérales à cet âge, et que les deux formes de céphalées sont souvent associées. La différence la plus importante est la survenue très rare de nausées et de vomissements lors des céphalées de tension [4, 10].
Le traitement consiste à mettre l’enfant au repos, et à lui administrer un traitement visant à arrêter l’accès migraineux : ibuprofène (10 mg/kg) en première intention, sumatriptan sous forme de spray nasal à partir de 12 ans en cas d’échec de l’ibuprofène. Le paracétamol est peu efficace et les autres triptans n’ont pas l’autorisation de mise sur le marché (AMM) en France avant l’âge de 18 ans [11].
Afin de réduire la fréquence des crises, il faut conseiller à l’enfant d’observer quelques règles d’hygiène de vie : suivre une alimentation équilibrée, avoir une activité physique modérée régulière et un sommeil régulier. Il est conseillé d’éviter les facteurs déclenchants des crises lorsqu’ils sont identifiés. Les psychothérapies comme la relaxation, le rétrocontrôle et la thérapie cognitivo- comportementale peuvent également être indiquées [11].
Si, malgré ces mesures, la migraine reste invalidante avec un nombre de crises supérieur à six par mois, ou à deux par semaine de façon régulière, un traitement de fond médicamenteux peut être proposé. Les principaux traitements ayant démontré leur efficacité sont : la flunarizine, le propanolol et le pizotifène. Ces traitements de fond sont donnés pour une durée d’au moins 3 mois, prolongée ou renouvelée en fonction de l’évolution des accès migraineux [11, 12].
L’hypertension intracrânienne peut être secondaire à une tumeur cérébrale, une hydrocéphalie, une hypertension intracrânienne idiopathique. Les céphalées sont le signe clinique le plus fréquent chez le grand enfant (voir chapitre 22.4), alors que chez le petit enfant (de moins de 5 ans) d’autres signes d’HTIC comme des vomissements ou une augmentation du périmètre crânien sont au premier plan [13]. Le fond d’oeil recherche un oedème papillaire mais son absence ne permet pas d’exclure le diagnostic [14]. Typiquement, les céphalées d’HTIC apparaissent progressivement, sont quotidiennes et plus intenses le matin, diffuses, s’accompagnent de nausées, vomissements, s’aggravent lors de la toux, des efforts physiques, de la manoeuvre de Valsalva. Les éclipses visuelles sont fréquentes. Cette symptomatologie typique d’HTIC aiguë nécessite la réalisation d’une imagerie cérébrale en urgence. En cas de céphalées chroniques, les indications d’imagerie cérébrale chez l’enfant sont : des signes cérébelleux (ataxie, nystagmus, tremblement d’intention), des signes d’HTIC (un oedème papillaire de stase, une augmentation du périmètre crânien), un déficit neurologique focal, un changement de personnalité de l’enfant, une baisse des résultats scolaires [15]. L’imagerie cérébrale permet d’identifier un processus expansif intracérébral, une dilatation ventriculaire en cas d’hydrocéphalie, des signes indirects d’hypertension intracrânienne idiopathique (aspect de selle turcique vide, aplatissement postérieur des globes oculaires, distension de la gaine des nerfs optiques) et d’éliminer une thrombose veineuse cérébrale. La mesure de la pression du liquide céphalorachidien confirme le diagnostic d’HTIC idiopathique si elle est supérieure à 28 cm d’eau chez l’enfant.
Il s’agit des céphalées réfractives ou attribuées à des hétérophories décompensées, survenant lors des efforts visuels prolongés chez les enfants d’âge scolaire. Elles s’accompagnentd’une sensation de brouillard visuel, et parfois d’épisodes de diplopie binoculaire transitoires. La prescription d’une correction optique adaptée, après mesure de la réfraction sous cycloplégie, peut les améliorer significativement.
Pour pouvoir être rattachées au traumatisme crânien, les céphalées doivent survenir dans les 7 jours qui suivent le traumatisme. Elles peuvent être aiguës si leur durée est inférieure à 3 mois ou chroniques. Elles sont une plainte fréquente après un traumatisme crânien, n’ont pas de caractéristiques cliniques qui leur sont propres, et leur intensité n’est pas proportionnelle à la gravité du traumatisme. C’est l’association à d’autres signes cliniques à l’examen neurologique qui nécessite de réaliser une imagerie cérébrale, qui retrouverait des lésions traumatiques cérébrales dans moins de 10 % des cas après un traumatisme crânien mineur chez l’enfant [16].
[1] Genizi J, Khourieh Matar A, Zelnik N, et al. Frequency of pediatric migraine with aura in a clinic-based sample. Headache 2016 ; 56 : 113-7.
[2] Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia 2013 ; 33 : 629-808.
[3] Muttamthottil Varghese F. Brief migraine episodes in children and adolescents-a modification to International Headache Society pediatric migraine (without aura) diagnostic criteria. Springerplus. 2013 ; 2 : 77.
[4] Özge A, Yalın OÖ. Chronic migraine in children and adolescents. Curr Pain Headache Rep 2016 ; 20 : 14.
[5] McAbee GN, Morse AM, Assadi M. Pediatric Aspects of Headache Classification in the International Classification of HeadacheDisorders-3 (ICHD-3 beta version). Curr Pain Headache Rep 2016 ; 20 : 7.
[6] He Y, Li Y, Nie Z. Typical aura without headache : a case report and review of the literature. J Med Case Rep 2015 ; 9 : 40.
[7] Teixeira KC, Montenegro MA, Guerreiro MM. Migraine equivalents in childhood. J Child Neurol 2014 ; 29 : 1366-9.
[8] Lagman-Bartolome AM, Lay C. Pediatric migraine variants : a review of epidemiology, diagnosis, treatment, and outcome. Curr Neurol Neurosci Rep 2015 ; 15 : 34.
[9] Rothner AD, Parikh S. Migraine variants or episodic syndromes that may be associated with migraine and other unusual pediatric headache syndromes. Headache 2016 ; 56 : 206-14.
[10] Lütschg J. Les céphalées primaires de l’enfant: diagnostic et traitement. Paeditrica 2007 ; 18 : 39-44.
[11] Cuvellier JC. Céphalées et migraines de l’enfant. Encycl Méd Chir (Elsevier, Paris). Traité de médecine, 2013 : 8 p.
[12] Parain D, Mihl M. Phénomènes paroxystiques non épileptiques. In : Chabrol B, Dulac O, Mancini J, Ponsot G. Eds. Neurologie pédiatrique. 3e éd. Médecine-Sciences- Flammarion ; 2010, p. 382-89.
[13] Chu TP, Shah A, Walker D, Coleman MP. Pattern of symptoms and signs of primary intracranial tumours in children and young adults : a record linkage study. Arch Dis Child 2015 ; 100 : 1115-22.
[14] Chelse AB, Epstein LG. Intracranial hypertension in children without papilledema. Pediatr Neurol Briefs 2015 ; 29 : 61.
[15] Abu-Arafeh I, Macleod S. Serious neurological disorders in children with chronic headache. Arch Dis Child 2005 ; 90 : 937-40.
[16] Chelse AB, Epstein LG. Blunt head trauma and headache. Pediatr Neurol Briefs 2015 ; 29 : 30.