Pathologie du vitré
Coordonné par P. Gastaud
E. N’Guyen, P Gastaud
À la 5e semaine de vie embryonnaire, la placode cristallinienne s’invagine pour former la fossette cristallinienne. La placode se détache de l’ectoderme superficiel et devient la vésicule cristallinienne, interceptée par la cupule optique. Le corps vitré primitif, matrice gélatineuse constituée de matériel fibrillaire, apparaît entre la vésicule cristallinienne et la paroi interne de la cupule optique : l’espace lenticulorétinien. La vésicule cristallinienne et la rétine sont vascularisées par l’artère hyaloïdienne qui est une branche terminale de l’artère ophtalmique. Cette artère hyaloïdienne relie la papille à la face postérieure du cristallin et donne des branches qui forment un lacis vasculaire dans le vitré primitif.
À la fin de la 6e semaine de vie embryonnaire, les premières fibres du vitré secondaire apparaissent, formées par les hyalocytes. Pendant ce temps, le pédoncule optique se replie autour de son axe ventral, enfermant ainsi l’artère hyaloïdienne dans la fente colobomique qui se ferme.
Au 4e mois, le cristallin perd la nécessité d’être vascularisé et la portion de l’artère qui traverse le corps vitré pour atteindre le cristallin dégénère par des processus apoptotiques et nécrotiques, alors que le reste deviendra l’artère centrale de la rétine. Au 7e mois, le flux sanguin dans le système hyaloïdien s’interrompt complètement et les vaisseaux régressent. Un canal hyaloïdien déshabité relie ainsi la papille à la face postérieure du cristallin : il s’agit du canal de Cloquet. Le vitré secondaire en place est alors avasculaire et constituera le vitré définitif.
La régression du système hyaloïdien primitif est guidée par des processus apoptotiques et nécrotiques [1]. Il semblerait que l’arrêt de la sécrétion du facteur de croissance endothélial vasculaire (vascular endothelial growth factor ou VEGF) provoquerait l’apoptose [2]. Les hyalocytes présents dans le vitré auraient une action similaire aux phagocytes mononucléaires et pourraient participer à la dégénérescence du vitré primaire [1].
La persistance du système vasculaire hyaloïdien est le résultat d’une résorption incomplète du système hyaloïdien. Les présentations cliniques sont nombreuses et très variables en fonction du degré de non-résorption. L’échographie Doppler permet d’objectiver ces vestiges embryonnaires avec mesure du flux sanguin. On peut observer à l’âge adulte des vestiges de ce réseau vasculaire sous différentes formes, comme notamment : un fin reliquat artériel flottant dans le vitré ; la tache de Mittendorf qui est un reliquat vasculaire observé au niveau de la capsule postérieure du cristallin, habituellement en inféronasal ; la papille de Bergmeister qui est un reliquat fibroglial situé en avant de la tête du nerf optique ; un véritable cordon fibroglial dense vascularisé reliant le nerf optique à la face postérieure du cristallin ; les kystes vitréens qui sont des kystes situés dans le vitré, contenant des résidus vasculaires hyaloïdiens.
L’impact fonctionnel dépend du degré d’involution du système vasculaire hyaloïdien. Les vestiges mineurs peuvent être découverts fortuitement lors d’un examen de routine, alors que les reliquats majeurs sont susceptibles de provoquer des amblyopies ou des complications hémorragiques [1].
La persistance de la vascularisation foetale (PVF) est une anomalie de la résorption du système vasculaire hyaloïdien. Il s’agit d’une dysembryopathie oculaire rare, décrite par Reese [1], affectant des enfants nés à terme et en bonne santé. L’atteinte est habituellement sporadique. Cependant, quelques cas familiaux ont été rapportés, avec notamment la mise en évidence d’un locus sur le chromosome 10q11 au sein d’une famille pakistanaise consanguine atteinte sur six générations [2]. Cette affection est le plus souvent unilatérale, indépendante du sexe et de la latéralité. Les formes bilatérales sont plus rares et doivent faire rechercher des maladies systémiques et neurologiques.
La leucocorie, le strabisme et la microphtalmie sont les signes révélateurs les plus fréquents, rendant le diagnostic assez précoce. Plus le degré de sévérité est élevé, plus le diagnostic est fait précocement. Une anesthésie générale est souvent nécessaire pour pratiquer un examen ophtalmologique complet et précis. Trois formes cliniques peuvent être observées :
la forme antérieure ; on peut observer de manière inconstante les éléments suivants : une microphtalmie, une microcornée, une leucocorie liée à une cataracte capsulaire postérieure et à une membrane fibrovasculaire rétrolentale. Celle-ci est de taille variable et se manifeste par soit une petite opacité isolée à la face postérieure du cristallin (tache de Mittendorf, Fig. 15-1), soit une large membrane vascularisée rétrocristallinienne étendue contre la capsule postérieure (Fig. 15-2). Elle peut avoir des anastomoses avec les vaisseaux iriens et entraîner en se contractant un étirement centripète des procès ciliaires (Fig. 15-3). Les formes antérieures sont probablement sous-évaluées, car elles sont considérées comme des cataractes congénitales [3] ;
la forme postérieure : elle est plus rare. La papille de Bergmeister est un reliquat de fibrose prépapillaire ; elle constitue une forme minime de PVF. On pourra observer la persistance d’une artère hyaloïdienne, adhérant à la papille. Des formes plus sévères peuvent être constatées, telles que les condensations vitréennes prérétiniennes, les tractions vitréennes, les décollements de rétine tractionnels ainsi que les plis falciformes [3] ;
la forme mixte : elle est la plus fréquente. Il s’agit de l’association d’une membrane fibrovasculaire rétrolentale et d’un cordon vasculaire hyaloïdien reliant le cristallin à la papille (Fig. 15-4 et 15-5).
Une échographie type B peut être nécessaire, surtout si la membrane vasculaire rétrolentale gêne l’accès au fond d’oeil. Elle permet d’éliminer la présence de calcifications intra-oculaires et de mettre
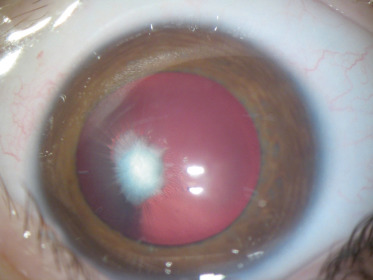
Fig. 15-1 Opacité à la face postérieure du cristallin correspondant à une tache de Mittendorf.
(Remerciements au Dr P. Dureau.)
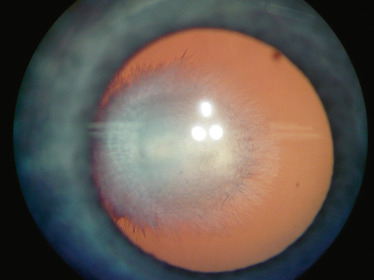
Fig. 15-2 Large membrane vascularisée rétrocristallinienne étendue contre la capsule postérieure.
(Remerciements au Dr P. Dureau.)
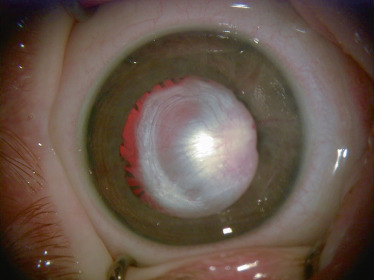
Fig. 15-3 Volumineuse membrane fibrovasculaire rétrolentale ayant des anastomoses avec les vaisseaux iriens, entraînant un étirement centripète des procès ciliaires par contraction de la membrane.
(Remerciements au Dr P. Dureau.)
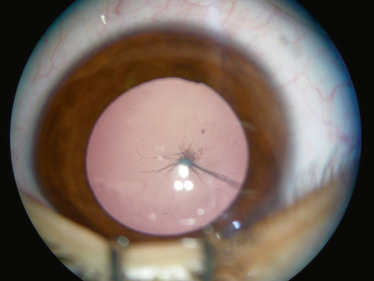
Fig. 15-4 Opacité cristallinienne postérieure reliée à un cordon fibrovasculaire tendu vers le pôle postérieur.
(Remerciements au Dr P. Dureau.)
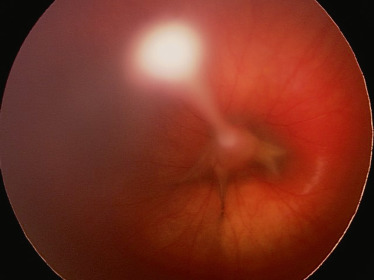
Fig. 15-5 Cordon fibrovasculaire reliant la face postérieure du cristallin à la papille.
(Remerciements au Dr P. Dureau.)
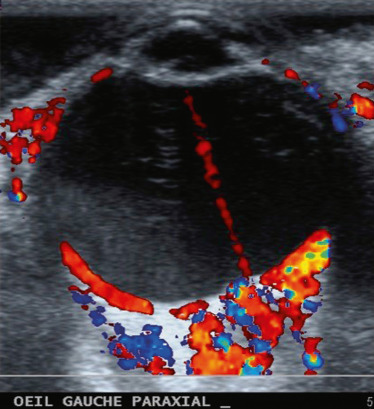
Fig. 15-6 Échographie Doppler objectivant la présence d’une artère hyaloïdienne persistante, avec la mise en évidence d’un flux sanguin.
(Remerciements au Dr P. Dureau.)
en évidence un décollement de rétine (DR) tractionnel. Elle permet également de mesurer la longueur axiale du globe oculaire, notamment en cas de microphtalmie. Le flux sanguin persistant peut être apprécié par une échographie Doppler (Fig. 15-6). Une tomodensitométrie et/ou une imagerie par résonance magnétique (IRM) cérébrale peuvent être réalisées afin de confirmer les données ci-dessus et d’éliminer d’autres anomalies malformatives cérébrales.
Les PVF sont souvent stables. Des complications rares peuvent cependant survenir :
hémorragies intravitréennes ou intracapsulaires (Fig. 15-7) ;
poussées inflammatoires par effraction de la capsule cristallinienne ;
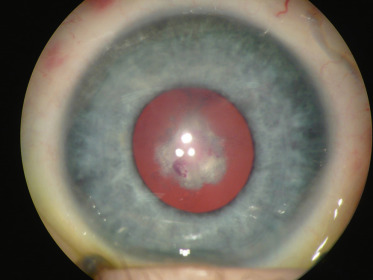
Fig. 15-7 Complication hémorragique intracapsulaire d’une membrane fibrovasculaire rétrocristallinienne.
(Remerciements au Dr P. Dureau.)
poussées d’hypertonie intra-oculaire douloureuses soit par intumescence cristallinienne, soit par prétraction de la membrane fibrovasculaire à partir des procès ciliaires avec bascule antérieure de l’ensemble iridocristallinien, provoquant une athalamie et un blocage de l’angle iridocornéen. Un décollement ciliaire par étirement des procès ciliaires visibles dans l’aire pupillaire peut entraîner une hypotonie puis évoluer vers une phtyse.
Enfin, ces anomalies congénitales sont des facteurs de risque majeur d’amblyopie organique.
La PVF peut être associée à une microphtalmie (critère de mauvais pronostic), un colobome choriorétinien, un colobome du nerf optique. Certaines équipes ont rapporté d’autres anomalies oculaires comme des cas de morning glory [4], des anomalies de Peters ou encore des kystes rétiniens.
Les associations systémiques sont habituellement observées dans les formes de PVF bilatérales. Des cas de trisomie 13 et 15 [5] ont été rapportés dans la littérature. De rares associations avec d’autres pathologies ont été décrites, telles que le syndrome d’Aicardi [6] et la neurofibromatose de type 2 [7].
Certains diagnostics différentiels sont à évoquer et doivent être écartés avec précaution. Le rétinoblastome doit être éliminé par une échographie ou une imagerie cérébrale avec la recherche de microcalcifications intra-oculaires. Une PVF peut être confondue avec une cataracte congénitale isolée : il est indispensable d’éliminer une vascularisation foetale par une échographie Doppler. Pour les formes postérieures, les diagnostics différentiels sont les décollements de rétine congénitaux qui restent exceptionnels, les rétinopathies des prématurés dont le contexte est différent et les maladies de Coats. La maladie de Norrie peut se manifester par une leucocorie, avec une cataracte et une dysplasie rétinienne. Cependant, l’atteinte est souvent bilatérale et la transmission est liée à l’X. De plus, la maladie de Norrie associe habituellement la dysplasie rétinienne à un retard mental et une perte auditive de perception. L’échographie Doppler, la tomodensitométrie, ainsi que l’IRM cérébrale aident à éliminer ces différents diagnostics.
Le pronostic fonctionnel dépend de la forme clinique : les formes postérieures et les formes mixtes sont de moins bon pronostic que les formes antérieures. Le degré d’atteinte de l’axe visuel conditionne le pronostic visuel. Le délai diagnostique ainsi que le délai chirurgical influencent également ce pronostic : un traitement chirurgical tardif augmente considérablement le risque d’amblyopie organique. De plus, la présence d’une microphtalmie et d’autres anomalies oculaires congénitales sont de mauvais pronostic.
La prise en charge dépendra du degré de sévérité de l’affection et de l’association ou non à d’autres malformations oculaires. Les formes antérieures obturantes à fort risque amblyogène bénéficient d’une chirurgie précoce. En revanche, les indications chirurgicales sont controversées dans les formes postérieures ou mixtes, car la récupération visuelle est plus incertaine [8]. Si un traitement chirurgical peut être envisagé, il est préconisé d’opérer le plus précocement possible afin de diminuer le risque d’amblyopie. Certaines équipes proposent une chirurgie à visée esthétique ou bien pour éviter les complications glaucomateuses ou hémorragiques, mais aucunes recommandations n’ont été clairement établies à ce jour.
Les prises en charge seront discutées au cas par cas, mais de manière générale, une abstention chirurgicale sera préférée pour les formes antérieures non obturantes et les formes postérieures minimes. Une correction optique adaptée ainsi que le traitement d’une éventuelle amblyopie restent indispensables.
exceptionnelles. Les kystes congénitaux seraient issus du système vasculaire hyaloïdien [9]. Ils sont généralement localisés dans le vitré postérieur et peuvent être fixés au nerf optique par un cordon fibrovasculaire. Asymptomatiques, ils sont habituellement isolés, avec l’absence de pathologie intra-oculaire. Ils sont stables et évoluent peu au fil du temps. Les analyses anatomopathologiques des kystes vitréens ont révélé une architecture monocellulaire épithéliale prédominante contenant des mélanosomes, compatible avec un choristome kystique issu du système hyaloïdien [9].
Il semblerait que certains kystes pigmentés seraient issus de l’épithélium pigmentaire irien. Ces kystes iriens disloqués libres sont très rares. Shields et al. ont proposé une classification des kystes iriens en fonction de leur localisation [10]. Dans leur série de 53 kystes iriens constatés chez des enfants [11], 59 % étaient périphériques (au niveau de la racine de l’iris), 9 % étaient intermédiaires (entre le bord pupillaire et la racine de l’iris), 5 % étaient centraux (pupillaires), et 4 % étaient détachés (flottant librement) en chambre antérieure ou dans la cavité vitréenne. Ces kystes iriens libres flottent dans le vitré (Fig. 15-8) [6], et suivent les mouvements oculaires [10].
En outre, des kystes vitréens acquis ont été décrits. Des cas de kystes post-traumatiques ont été rapportés : il s’agirait de kystes iriens préexistants qui se détacheraient et deviendraient symptomatiques à la suite du traumatisme. Il y a également des kystes secondaires à une chirurgie de DR [12] ou encore des kystes associés à des rétinites pigmentaires [13]. D’autres sont observés dans le cadre d’une inflammation intr-aoculaire telles que la toxoplasmose ou la cysticercose, où la tête du parasite peut être visible sur les parois du kyste [14].
Le diagnostic des kystes vitréens est établi au fond d’oeil biomicroscopique. L’échographie mode B retrouve une formation arrondie à parois hyperéchogènes et à contenu liquidien (Fig. 15-9). La tomographie par cohérence optique montre un kyste vitréen avec une zone d’ombre de la rétine en regard, sans arrêt complet du signal étant donné la finesse de la paroi kystique (Fig. 15-10).
Les kystes vitréens sont habituellement asymptomatiques, de découverte fortuite et ne nécessitent donc aucune prise en charge thérapeutique. Toutefois si le patient est gêné par la topographie trop axiale, un traitement peut être proposé. Certaines équipes ont procédé à une photodisruption au laser argon ou yttrium aluminium garnet (YAG) [15]. Sinon, une vitrectomie avec ablation du kyste peut être proposée [9].
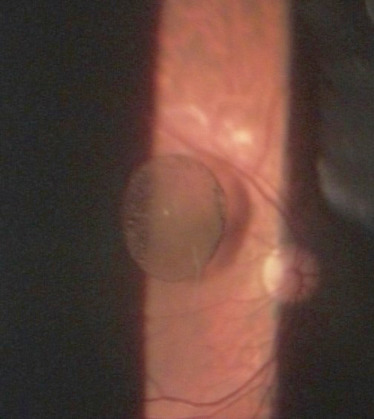
Fig. 15-8 Photographie à la lampe à fente : kyste vitréen pigmenté flottant dans le vitré postérieur, relié à un petit cordon fibreux..
(Remerciements au Dr C. Franceschetti et au Dr P. Gastaud.)
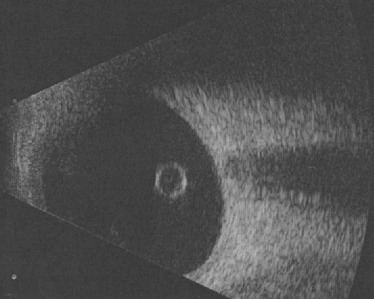
Fig. 15-9 Échographie mode B : kyste vitréen à parois hyperéchogènes flottant devant la papille.
(Remerciements au Dr C. Franceschetti.)
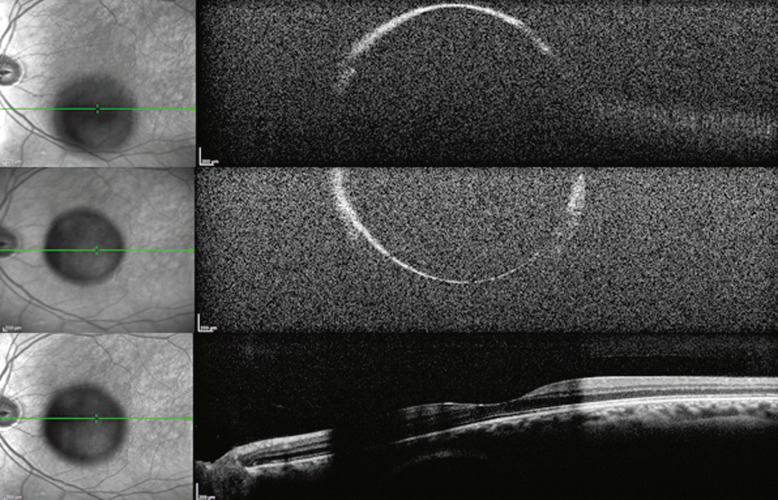
Fig. 15-10 Kyste vitréen flottant dans le vitré postérieur, en regard de la région maculaire.
La tomographie par cohérence optique (optical coherence tomography [OCT]) montre l’ombre du kyste sur la macula, sans arrêt complet du signal étant donné la finesse des parois kystiques.
(Remerciements au Dr C. Franceschetti.)
[1] Reese AB, Payne F. Persistence and hyperplasia of the primary vitreous : tunica vasculosa lentis or retrolental fibroplasia. Trans Am Ophthalmol Soc 1945 ; 43 : 163-92.
[2] Khaliq S, Hameed A, Ismail M, et al. Locus for autosomal recessive nonsyndromic persistent hyperplastic primary vitreous. Invest ophthalmol Vis Sci 2001 ; 42 : 2225-8.
[3] Bosjolie A, Ferrone P. Visual outcome in early vitrectomy for posterior persistent fetal vasculature associated with traction retinal detachment. Retina 2015 ; 35 : 570-6.
[4] Fei P, Zhang Q, Li J, Zhao P. Clinical characteristics and treatment of 22 eyes of morning glory syndrome associated with persistent hyperplastic primary vitreous. Br J Ophthalmol 2013 ; 97 : 1262-7.
[5] Iwig M. Retinal dysplasia. Morphological identity of the retinal dysplasia syndrome (Reese) and D (13-15) trisomy syndrome (Patau). Zentralbl Allg Pathol 1969 ; 112 : 492- 504.
[6] Beby F, Zech C, Touraine R, et al. Persistent hyperplastic primary vitreous syndrome in a girl with Aicardi syndrome. J Fr Ophtalmol 2000 ; 23 : 703-7.
[7] Nguyen DQ, Chatterjee S, Bates R. Persistent hyperplastic primary vitreous in association with neurofibromatosis 2. J Pediatr Ophthalmol Strabismus 2005 ; 42 : 247-9.
[8] Gastaud P, Costet C. Anomalies du développement. In : Brasseur G. Pathologie du vitré. Rapport SFO 2003. Paris : Masson ; 2003, p. 119-32.
[9] Nork TM, Millecchia LL. Treatment and histopathology of a congenital vitreous cyst. Ophthalmology 1998 ; 105 : 825-30.
[10] Shields JA, Kline MW, Augsburger JJ. Primary iris cysts : a review of the literature and report of 62 cases. Br J Ophthalmol 1984 ; 68 : 152-66.
[11] Shields JA, Shields CL, Lois N, Mercado G. Iris cysts in children : classification, incidence, and management. The 1998 Torrence A Makley Jr Lecture. Br J Ophthalmol 1999 ; 83 : 334-8.
[12] Asiyo-Vogel MN, el-Hifnawi el S, Laqua H. Ultrastructural features of a solitary vitreous cyst. Retina 1996 ; 16 : 250-4.
[13] Wagenaar JW. Vitreous cyst with retinitis pigmentosa ; a new syndrome ? Br J Ophthalmol 1952 ; 36 : 492-8.
[14] Saracco JB, Mouly A, Estachy G, et al. Vitreous cysticercosis : report of a case. Med Trop 1982 ; 42 : 97-9.
[15] Ruby AJ, Jampol LM. Nd:YAG treatment of a posterior vitreous cyst. Am J Ophthalmol 1990 ; 110 : 428-9.
F. Amouyal , B. Butet, C. Landré, F. Matonti , F. Metge-Galatoire , P. Gastaud
F. Amouyal, F. Matonti
Les plis rétiniens congénitaux et, plus précisément, les plis papillomaculaires touchent des sujets présentant des hypermétropies extrêmes, le plus souvent supérieures à 10 dioptries, et sont à l’origine d’amblyopies organiques. Ils résultent de l’asynergie entre une coque sclérale de volume réduit et un contenu choriorétinien en excès relatif créant ainsi une redondance tissulaire (Fig. 15-11). Le premier cas a été décrit en 1935 par Mann [1], qui nomma cette anomalie « ligament falciforme maculaire » .
La découverte peut être fortuite ou dans le cadre d’un dépistage familial. Les plis rétiniens congénitaux rentrent souvent dans le cadre d’une histoire familiale puisque sur 50 cas observés par Nishina et al., 64 % avaient un membre de la famille atteint [2]. Les symptômes pouvant être retrouvés sont un mauvais comportement visuel et/ou une faible fixation, un nystagmus, un strabisme ou une leucocorie. Nishina, dans sa série publiée de 147 yeux, montre que ces plis sont unilatéraux dans 78 % des cas, mais que 70 % de ces yeux peuvent être associés à une anomalie vasculaire rétinienne de l’oeil adelphe. Les plis se localisent en temporal dans 92 % des cas. L’acuité visuelle est supérieure à
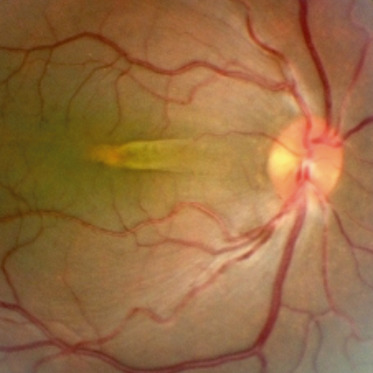
Fig. 15-11 Pli rétinien congénital.
20/100 dans seulement 4 % des cas, supérieure à 20/200 dans 41 % des cas et enfin, inférieure à 20/200 dans 59 % des cas. Une des causes largement décrite dans la littérature est représentée par la microphtalmie postérieure avec plis papillomaculaires, pathologie rare et le plus souvent isolée, sans atteinte systémique associée (à l’inverse de la microphtalmie congénitale) [3]. Elle se distingue de cette dernière par le fait que le diamètre cornéen, la chambre antérieure et le cristallin sont de dimensions normales, alors qu’ils sont diminués dans la microphtalmie congénitale. Elle associe un segment postérieur rétréci aboutissant à des plis rétiniens surélevés et bilatéraux, une hypermétropie souvent supérieure à 12 dioptries, ainsi qu’une acuité visuelle basse [4, 5]. Une amblyopie organique peut survenir en fonction de la localisation maculaire du pli.
Dans la microphtalmie postérieure, Boynton et al. [3] avaient émis l’hypothèse qu’un arrêt de la croissance des couches externes postérieures de l’oeil serait responsable d’un segment postérieur court. Ceci contrasterait avec une rétine neurosensorielle qui, elle, aurait continué sa formation aboutissant à la formation du typique pli papillomaculaire.
Fried et al. [6] ont étudié ces yeux microphtalmes en échographie et ont été les premiers à documenter ces plis papillomaculaires. Plus récemment, Jackson et al. [7] ont décrit ces anomalies en spectral-domain optical coherence tomography (SDOCT) et ont suggéré une possible transmission ou prédisposition génétique en raison de la similitude des anomalies retrouvées : les plis rétiniens n’impliquaient que les couches internes et épargnaient la membrane limitante externe et les photorécepteurs.
Dans la littérature, des plis rétiniens ont été décrits en association avec de nombreuses pathologies :
la vitréorétinopathie exsudative familiale (VREF), dysplasie vitréorétinienne génétique décrite ci-dessous ;
la rétinopathie du prématuré (retinopathy of prematurity ou ROP) : elle peut aboutir dans ses stades évolués à la formation de plis rétiniens, notamment en raison du tissu fibrovasculaire qui, après avoir proliféré à la jonction rétine vascularisée/rétine ischémique, se rétracte progressivement et forme ainsi un pli ;
la persistance primitive hyperplasique du vitré : elle a été décrite comme pouvant être associée à un pli rétinien congénital partant de la papille et adhérant au cristallin.
Les plis ont été aussi décrits dans le syndrome du bébé secoué, le rétinoschisis lié à l’X, le syndrome de Walker-Warburg, l’incontinentia pigmenti, la maladie de Norrie et dans les pathologies infectieuses choriorétiniennes.
Les complications potentielles de ces plis congénitaux peuvent être l’apparition d’un DR tractionnel ou rhegmatogène, une prolifération fibrovasculaire à partir d’une néovascularisation prérétinienne ou un DR exsudatif.
Il comprend, dans les formes secondaires, différentes modalités comme la photocoagulation au laser argon de zones ischémiques ou la cryoapplication de zones néovascularisées périphériques, la vitrectomie avec pelage de membranes épirétiniennes, un cerclage laser (± gaz) en fonction des complications associées.
En conclusion, malgré un diagnostic et un traitement précoce chez le jeune enfant (laser et chirurgie vitréorétinienne), la présence de plis rétiniens congénitaux entraîne très souvent un pronostic fonctionnel sombre ; un dépistage et un suivi de l’oeil adelphe et de la fratrie doivent systématiquement y être associés.
B. Butet
Le syndrome de Walker-Warburg (SWW), également nommé syndrome d’hydrocéphalie-agyrie-dysplasie rétinienne, est une forme rare de dystrophie musculaire congénitale (DMC) associée à des anomalies cérébrales et oculaires. Les DMC sont un groupe très hétérogène de myopathies d’origine génétique, majoritairement autosomiques récessives et d’un grand polymorphisme clinique.
La distribution du SWW est mondiale. L’incidence globale de cette forme de DMC n’est pas connue, mais une étude italienne a rapporté un taux d’incidence de 1,2 pour 100 000 naissances [8].
Ce syndrome appartient au groupe des dystroglycanopathies. L’α- dystroglycane est une protéine de la membrane externe de la membrane basale des cellules musculaires squelettiques, elle est impliquée dans l’ancrage des fibres musculaires à la matrice cellulaire externe. On la retrouve également dans certaines structures de l’oeil et du cerveau. Il existe une grande variabilité génétique du SWW : six gènes ont été identifiés à ce jour (POMT1, POMT2, POMGnT1, FKRP, FCMD, LARGE). La transmission est autosomique récessive.
Le SWW [9, 10] est défini par l’association d’une dystrophie musculaire congénitale et d’anomalies cérébrales et oculaires. Le tableau est sévère dès la naissance.
L’atteinte musculaire se caractérise par une hypotonie avec faiblesse musculaire diffuse, notamment des troubles majeurs de la déglutition et de la respiration. Une encéphalocèle et une hydrocéphalie sont souvent présentes et il existe un retard de développement global important. Sont décrits mais de façon inconstante : malformation de Dandy-Walker, atteinte des organes génitaux externes, fente labiopalatine, dysmorphie faciale.
Sur le plan oculaire, une microphtalmie uni- ou bilatérale est très fréquente, avec d’importantes anomalies du segment antérieur (anomalie de Peters, cataracte congénitale, colobomes) souvent responsables d’un glaucome congénital secondaire ; une buphtalmie peut être présente dans ce cas. Le fond d’oeil retrouve une dysplasie vitréorétinienne majeure avec persistance de la vascularisation foetale, une rétine pâle, des anomalies vasculaires rétiniennes et choroïdiennes, une absence de reflet fovéolaire. On peut noter des hémorragies vitréennes et/ou rétiniennes, un DR, un colobome ou une hypoplasie du nerf optique [11].
L’IRM cérébrale est capitale, elle révèle une lissencéphalie (malformation du cortex cérébral caractérisée par une absence congénitale des circonvolutions cérébrales) de type 2, éventuellement associée à des anomalies de signal de la substance blanche, une fusion des lobes occipitaux ou des malformations cérébelleuses. Des kystes arachnoïdiens au niveau de la fosse postérieure peuvent être présents, et parfois des méningo- ou encéphalocèles [11]. Au niveau des globes oculaires, l’IRM peut montrer par ailleurs des hypo- ou hypersignaux oculaires compatibles avec une persistance du vitré primitif, une hémorragie intravitréenne ou un DR. Ces lésions oculaires peuvent être facilement explorées par une échographie oculaire en mode B.
Les biopsies musculaires (avec analyse immunohistochimique) confirment la dystroglycanopathie. La biologie montre un taux élevé de créatine kinase.
Il s’agit des autres pathologies du groupe des dystroglycanopathies qu’il n’est pas toujours aisé de différencier, car il existe un continuum clinique entre toutes ces formes avec un spectre d’atteintes assez large et un ensemble de mutations génétiques similaires, d’expression phénotypique variable. Le SWW est la forme la plus sévère de dystrophie musculaire congénitale ; la plupart des enfants décèdent avant l’âge de 3 ans, souvent de défaillance respiratoire. Il n’y a pas de traitement spécifique, la prise en charge est palliative et préventive.
C. Landré
Le syndrome d’ostéoporose-pseudogliome est caractérisé par l’association d’une atteinte visuelle et d’une fragilité osseuse durant l’enfance [11].
Sur le plan ophtalmologique, on retrouve une cécité au plus tard à l’âge de 25 ans, mais le plus souvent dès la naissance avec un tableau de fibroplasie rétrolentale. Les anomalies oculaires retrouvées sont une dysplasie vitréorétinienne congénitale, une microphtalmie, un DR pseudogliomateux et une phtyse [13].
L’atteinte osseuse débute dans l’enfance, au plus tard vers l’âge de 20 ans. C’est en général au moment de l’apparition des lésions osseuses que le syndrome d’ostéoporose-pseudogliome est évoqué. Le plus souvent, il s’agit de fractures apparues après des traumatismes mineurs. En outre, un retard mental modéré peut être présent de manière inconstante, ainsi qu’une obésité.
Il s’agit d’une maladie rare (1/2 000 000), se transmettant sur un mode autosomique récessif [12]. En 2001, Gong et al. ont été les premiers à décrire une mutation du gène LRP5 associée à cette pathologie. Il est intéressant de noter que les porteurs hétérozygotes présentent une densité minérale osseuse plus faible que la normale et que cette mutation a été retrouvée chez des patients atteints d’ostéoporose qualifiée d’idiopathique [14].
La chirurgie vitréorétinienne peut être tentée pour prendre en charge les anomalies oculaires mais malheureusement, le plus souvent, le seul bénéfice à en attendre est d’éviter la phtyse oculaire. Différents traitements de l’ostéoporose et notamment le biphosphonate sont à la disposition des rhumatologues pour la prise en charge de la fragilité osseuse et la prévention des fractures.
Les diagnostics différentiels comprennent les pathologies entraînant un DR bilatéral dans l’enfance et notamment la rétinopathie du prématuré, la vitréorétinopathie exsudative familiale et la persistance de la vascularisation foetale. Un autre diagnostic à éliminer dans ce type de situation clinique est le rétinoblastome bilatéral.
Le pronostic visuel est très sombre avec une cécité bilatérale rapportée au plus tard à l’âge de 25 ans. D’un point de vue rhumatologique, le pronostic est conditionné par la rapidité de la mise en place du traitement général.
C. Landré
Le syndrome de nanisme oculo-palato-cérébral ou oculo-palatocerebral dwarfism a été décrit pour la première fois en 1985 par Frydman et al. à propos d’une fratrie de trois enfants de parents consanguins. Les trois enfants présentaient l’association d’une microcéphalie, d’une petite taille, d’un retard mental et d’une persistance hypertrophique du vitré primitif [15].
À partir de la description des cinq cas rapportés (trois garçons et deux filles), des éléments cliniques constituant le syndrome ont pu être mis en évidence [15–17]. D’un point de vue ophtalmologique, une persistance hypertrophique du vitré primitif a été retrouvée chez tous les patients de manière uni- ou bilatérale. Une cataracte, une microphtalmie ou une atrophie du nerf optique étaient parfois associées. Le syndrome est caractérisé par l’association de quatre éléments : un retard mental, une microcéphalie, des anomalies du palais (le plus souvent à type de fente labiopalatine) et des anomalies oculaires. De manière inconstante, d’autres anomalies ont été décrites. Il s’agit tout d’abord d’anomalies prénatales avec une hypertension artérielle maternelle, un oligamnios ou un retard de croissance in utero (RCIU). Après la naissance, il existe le plus souvent un retard de croissance staturo-pondéral y compris des extrémités (mains et pieds). Les garçons présentent fréquemment une cryptorchidie. En outre, des anomalies squelettiques sont fréquentes et une atrophie cérébrale peut conduire à une quadriplégie. Enfin, on retrouve parfois des déficits auditifs et un terrain atopique.
Il s’agit d’une pathologie extrêmement rare, décrite uniquement chez cinq patients de trois familles différentes (dont deux familles consanguines). La transmission est donc supposée être autosomique récessive. Aucun gène n’a encore été retrouvé comme étant associé à cette pathologie.
F. Metge-Galatoire
La maladie de Norrie, décrite pour la première fois par Warburg en 1960 [18] est une maladie rare de transmission récessive liée à l’X qui touche à la fois l’oeil, l’oreille et le système nerveux central. Cette maladie est due à la mutation du gène NDP (Norrie disease pseudoglioma) et se caractérise par un développement rétinien anormal avec cécité congénitale bilatérale. Une surdité de perception et une atteinte du système nerveux central sont classiquement associées mais moins constantes et de révélation souvent plus tardive. La prévalence et l’incidence de la maladie ne sont pas connues en raison de sa rareté. On ne note aucune prédisposition ethnique.
Les signes oculaires, constamment bilatéraux, sont très précoces chez le nourrisson de sexe masculin dès les premières semaines de vie : une masse fibreuse rétrolentale est tout d’abord présente (historiquement appelée pseudogliome), très rapidement compliquée DR ; celui-ci est le plus souvent masqué par la prolifération fibreuse et diagnostiqué par l’échographie réalisée devant une leucocorie (Fig. 15-12a à d). Ce DR s’intègre dans le cadre d’une véritable dysplasie vitréorétinienne ; la structure histologique de la rétine y est anormale avec une stratification pathologique des couches cellulaires rétiniennes et une organisation en pseudo-rosettes [19]. L’iris, la chambre antérieure et la cornée peuvent être normales à la naissance mais une hypoplasie de l’iris, un ectropion de l’uvée, un étirement des procès ciliaires peuvent être observés. Au cours de l’évolution, et en l’absence de traitement, la prolifération fibrorétinienne se rétracte vers l’avant et conduit à un affaissement de la chambre antérieure avec risque de synéchies iridocristalliniennes (Fig. 15-12e) puis iridocornéennes, de kératite en bandelette et d’opacification cornéenne complète. Une microphtalmie est habituelle ; précoce, elle est associée à un défaut de croissance orbitaire.
Le déficit visuel va de la perception lumineuse à une cécité congénitale complète. Environ un tiers des garçons affectés développe une surdité de perception, asymétrique, progressive, qui n’est pas forcément présente à la naissance mais débute généralement dans l’enfance (âge médian d’apparition : 12 ans). La perte auditive peut être sévère et bilatérale à l’âge adulte. La moitié à deux tiers des patients présente un déficit intellectuel d’importance variable, pouvant aller, rarement, jusqu’à la psychose [20].
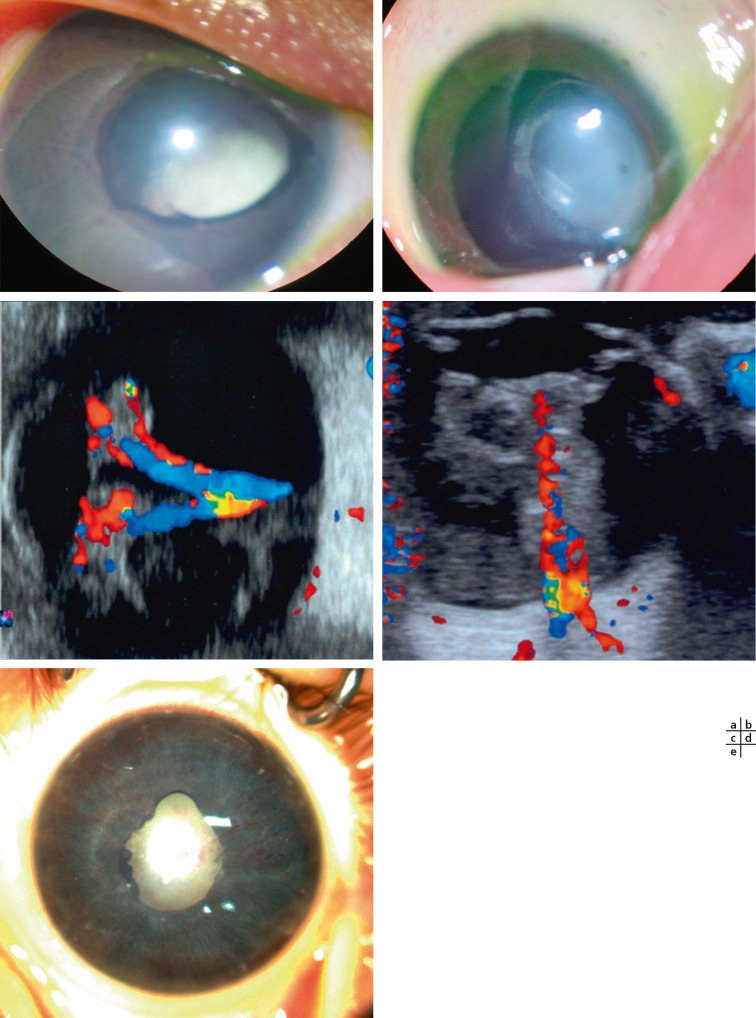
Fig. 15-12 Leucocorie chez des nourrissons atteints de maladie de Norrie.
a-d. Nourrisson de 5 mois : sur l’oeil droit (a), il existe un ectropion de l’uvée une dilatation des vaisseaux iriens. On visualise une prolifération fibreuse en arrière du cristallin qui est clair. Sur l’oeil gauche (b), on devine des vaisseaux rétiniens en arrière de la condensation rétrocristallinienne. L’échographie Doppler en couleurs montrant la rétine épaissie et totalement décollée au niveau des yeux droit (c) et gauche (d). e. Nourrisson de 4 mois : on note des synéchies iridocristalliniennes et une fibrose rétrolentale. La chambre antérieure est étroite avec un aspect d’iris tomate.
La Maladie de Norrie est due à des mutations du gène NDP (Xp11.4-p11.3). Ce gène cloné en 1992 est composé de 3 exons et code une protéine, la Norrin elle-même composée de 133 acides aminés et impliquée dans le développement neurologique et vasculaire de l’oeil, du cerveau et de l’appareil auditif [21, 22]. Cette protéine présente des domaines riches en cystéine similaires au transforming growth factor β (TGF-β) qui confèrent à la rétine ses propriétés structurelles [23]. L’identification du gène a permis le diagnostic génétique moléculaire et le dépistage anténatal de la maladie. Plus de 140 mutations ont été décrites dans la maladie de Norrie ou dans d’autres maladies rétiniennes dites « NDPdépendantes » que sont la persistance de la vascularisation foetale, la vitréorétinopathie exsudative familiale dans sa forme liée à l’X, certaines formes de rétinopathie des prématurés et de maladie de Coats (Tableau 15-1) [24–26]. Les formes de la maladie de Norrie associées à une délétion chromosomique incluant le locus NDP et les gènes des mono-amines oxydases A et B adjacents correspondent aux atteintes cliniques les plus graves sur le plan phénotypique ; dans ces cas, le retard mental est sévère et peut être associé à des mouvements anormaux, une démence et un hypogonadisme [27]. Les femmes conductrices sont en règle asymptomatiques mais de rares cas de manifestations à type de pli rétinien ou d’anomalies vasculaires périphériques ont été décrits [28]. Pour la majorité des patients, le séquençage direct du gène permet l’identification de la mutation responsable de la maladie. Lorsque la suspicion de la maladie ne peut pas être confirmée par le test du gène NDP, d’autres gènes, causant des conditions cliniques similaires de dysplasie rétinienne plus ou moins exsudative, doivent être analysés (FZD4, LRP5, TSPAN12, etc.).
Le diagnostic de la maladie repose sur les signes cliniques oculaires précoces caractéristiques (leucocorie bilatérale et DR précoce) et peut être confirmé par le test de génétique moléculaire du gène NDP. Celui-ci doit être fait systématiquement en cas de DR bilatéral chez un nourrisson de sexe masculin, après avoir éliminé le diagnostic de rétinoblastome en échographie et en l’absence de grande prématurité. En effet, les deux principaux diagnostics différentiels de la maladie sont le rétinoblastome dans sa forme bilatérale et la rétinopathie des prématurés de stade 5 (fibroplasie rétrolentale), facilement éliminée en l’absence de naissance prématurée. La vitréorétinopathie exsudative familiale doit également être évoquée, mais elle est le plus souvent asymétrique et le DR y est rarement aussi précoce. La PVF dans sa forme complète avec atteinte rétinienne, est quant à elle, exceptionnellement bilatérale. Un examen ORL sera également systématique pour dépister une surdité précoce, de même qu’un examen neuropédiatrique.
Un conseil génétique doit être systématiquement proposé aux parents d’un enfant atteint ; en cas de nouvelle grossesse, le test anténatal est possible par amniocentèse, lorsque la mutation à l’origine de la maladie a été identifiée dans la famille.

Un DR bilatéral est déjà présent le plus souvent au moment du diagnostic, et l’architecture rétinienne très anormale assombrit encore le pronostic visuel. Une chirurgie est néanmoins proposée pour éviter les complications trophiques (et l’énucléation qui en découle), en particulier lorsque le DR s’associe à une hypothalamie menaçante. La prise en charge chirurgicale comporte l’ablation du cristallin et de la rétraction fibreuse ; le DR se révèle le plus souvent difficile à disséquer étant donné les anomalies structurelles de la rétine. Cependant, le maintien d’une perception lumineuse positive chez ces enfants paraît plus fréquent en cas de chirurgie précoce par rapport à l’évolution naturelle [29].
P. Gastaud
Aussi appelé syndrome de Patau, elle est exceptionnelle. Elle associe une microphtalmie, une fente labiopalatine (50 % ), une polydactylie et une atteinte systémique. Les anomalies oculaires ont peu d’importance par rapport aux graves malformations cardiologiques et du système nerveux central, responsables d’un décès très précoce [30]. Le faciès est caractéristique (implantation basse des cheveux, nez bulbeux). Les anomalies ophtalmologiques les plus fréquentes sont microphtalmie, colobome de l’uvée, cataracte, hyperplasie et persistance du vitré primitif, dysplasie rétinienne.
[1] Mann I. Congenital retinal fold. Br J Ophthalmol 1935 ; 19 : 641-58.
[2] Nishina S, Suzuki Y, Yokoi T, et al. Clinical features of congenital retinal folds. Am J Ophthalmol 2012 ; 153 : 81-7.
[3] Boynton JR, Purnell EW. Bilateral microphthalmos without microcornea associated with unusual papillomacular retinal folds and high hyperopia. Am J Ophthalmol 1975 ; 79 : 820-6.
[4] Kida Y, Kurome H, Hayasaka S. Bilateral microphthalmos with poor visual acuity, high hyperopia, and papillomacular retinal folds in siblings. Jpn J Ophthalmol 1995 ; 39 : 177-9.
[5] Kiratli H, Tümer B, Kadayifçilar S. Bilateral papillomacular retinal folds and posterior microphthalmus : new features of a recently established disease. Ophthalmic Genet 2000 ; 21 : 181-4.
[6] Fried M, Meyer-Schwickerath G, Koch A. Excessive hypermetropia : review and case report documented by echography. Ann Ophthalmol 1982 ; 14 : 15-9.
[7] Jackson TE, Yang YC, Shun-Shin GA. Spectral domain optical coherence tomography findings in retinal folds associated with posterior microphthalmos. J AAPOS 2012 ; 16 : 389-91.
[8] Mostacciuolo ML, Miorin M, Martinello F, et al. Genetic epidemiology of congenital muscular dystrophy in a sample from north-east Italy. Hum Genet 1996 ; 97 : 277-9.
[9] Rivier F, Mercier M, Hugon G, et al. Dystrophies musculaires congénitales. Encycl Méd Chir (Elsevier, Paris). Neurologie, 17-173-A-20. 2010.
[10] Brasseur-Daudruy M, Vivier PH, Ickowick V, et al. Walker-Warburg syndrome diagnosed by findings of typical ocular abnormalities on prenatal ultrasound. Pediatric Radiology 2012 ; 42 : 488-90.
[11] Lee CY. Walker-Warburg syndrome : rare congenital muscular dystrophy associated with brain and eye abnormalities. Hong Kong Med J 2014 ; 20 : 556.e4-5
[12] Ai M, Heeger S, Bartels CF, Schelling DK. Clinical and molecular findings in osteoporosis- pseudoglioma syndrome. Am J Hum Genet 2005 ; 77 : 74153.
[13] Lee DH, Wenkert D, Whyte MP, et al. Congenital blindness and osteoporosis-pseudoglioma syndrome. J AAPOS 2003 ; 7 : 75-7.
[14] Gong Y, Slee RB, Fukai N, et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 2001 ; 107 : 513-23.
[15] Frydman M, Kauschansky A, Leshem I, Savir H. Oculo-palato-cerebral dwarfism : a new syndrome. Clin Genet 1985 ; 27 : 414-9.
[16] Pellegrino JE, Engel JM, Chavez D, Day-Salvatore D. Oculo-palatal-cerebral syndrome: a second case. Am J Med Genet 2001 ; 99 : 200-3.
[17] Alanay Y, Boduroglu K, Sönmez B, Orhan M. Oculo-palato-cerebral syndrome : a third case supporting autosomal recessive inheritance. Am J Med Genet 2004 ; 130A : 92-5.
[18] Warburg M. Norrie’s disease : a new hereditary bilateral pseudotumor of the retina. Acta Ophthalmol 1961 ; 39 : 757-72.
[19] Shroeder B, Hesse L, Brück W, Gal A. Histopathological and immunohistochemical findings associated with a null mutation in the Norrie disease gene. Ophthalmic Genet 1997 ; 18 : 71-7.
[20] Caputo G, Dureau P. Dysplasies vitréorétiniennes. In : Goberville M, Dureau P. Ophtalmologie pédiatrique et strabismes. Paris : Médecine sciences/Lavoisier ; 2014, p. 12-9.
[21] Sims KB, Lebo RV, Benson G, et al. The Norrie disease gene maps to a 150 kb region on chromosome Xp11.3. Hum Mol Genet 1992 ; 1 : 83-9.
[22] Urano T, Takamiya K, Furukawa K, Shiku H. Molecular cloning and functional expression of the second mouse nm23/NDP kinase gene, nm23-M2. FEBS Lett 1992 ; 309 : 358-62.
[23] Berger W, Meindl A, van de Pol TJ, et al. Isolation of a candidate gene for Norrie disease by positional cloning. Nat Genet 1992 ; 2 : 84.
[24] Berger W. Molecular dissection of Norrie disease. Acta anat (Basel) 1998 ; 162 : 95-100.
[25] Zhang XY, Jiang WY, Chen LM, Chen SQ. A novel Norrie disease pseudoglioma gene mutation, c.-1_2delAAT, responsible for Norrie disease in a Chinese family. Int J Ophthalmol 2013 ; 6 : 739-43.
[26] Sims KB. NDP-Related Retinopathies. In : GeneReviews® [Internet]. Pagon RA, Adam MP, Ardinger HH, et al. Eds. Seattle (WA) : University of Washington, Seattle 1993–2016. 1999 Jul 30 [updated 2014 Sep 18].
[27] Collins FA, Murphy DL, Reiss AL, et al. Clinical, biochemical, and neuropsychiatric evaluation of a patient with a contiguous gene syndrome due to a microdeletion Xp11.3 including the Norrie disease locus and monoamine oxidase (MAOA and MAOB) genes. Am J Med Genet 1992 ; 42 : 127-34.
[28] Lin P, Shankar SP, Duncan J, et al. Retinal vascular abnormalities and dragged maculae in a carrier with a new NDP mutation (c.268delC) that caused severe Norrie disease in the proband. J AAPOS 2010 ; 14 : 93-6.
[29] Walsh MK, Drenser KA, Capone A Jr, Trese MT. Early vitrectomy effective for Norrie disease. Arch Ophthalmol 2010 ; 128 : 456-60.
[30] Roche O. Altération du cristallin et de la zonule. In : Dufier JL, Kaplan J. OEil et génétique. Rapport SFO. Paris : Masson ; 2005, p. 187-211.
B. Butet, F. Matonti , P. Gastaud, P. Gascon, F. Metge-galatoire , C. Landré
Parmi les vitréorétinopathies héréditaires, maladies génétiques caractérisées par des anomalies rétiniennes et une dystrophie du vitré, nous développerons les plus fréquentes qui sont le syndrome de Stickler et le rétinoschisis juvénile. Ce groupe de maladie a longtemps été dominé par le « syndrome de Wagner-Stickler » ; aujourd’hui, on distingue la maladie de Wagner caractérisée par une atteinte uniquement oculaire et celle de Stickler plus fréquente qui associe au même tableau ophtalmologique d’autres anomalies extra-oculaires.
B. Butet
Au sein des membres d’une même famille suisse présentant une dégénérescence vitréorétinienne isolée, Wagner a décrit pour la première fois en 1938 ce syndrome clinique associant un vitré lacunaire « optiquement vide » à des anomalies rétiniennes et une atteinte du segment antérieur [1]. Pendant longtemps, cette maladie a été confondue avec le syndrome de Stickler. Les analyses cliniques de Maumenee [2] puis Graeminger [3], puis les études génétiques et la biologie moléculaire ont permis d’en avoir une meilleure compréhension et de séparer clairement les deux pathologies. C’est une maladie très rare, avec une prévalence estimée à 1/1 000 000. Cependant, du fait de l’expression variable des symptômes, elle est probablement sous-diagnostiquée.
La transmission se fait selon un mode autosomique dominant à pénétrance quasi complète et expressivité variable. La versicaneest une protéoglycane abondamment présente dans le tissu rétinien et le vitré et joue un rôle important dans son organisation structurelle en se liant aux molécules d’acide hyaluronique. Le gène codant (anciennement chondroitin sulfate proteoglycan 2 ou CSPG2, maintenant nommé VCAN) a été localisé sur le bras long du chromosome 5 (5q13-14). Différentes mutations ont été découvertes ces dernières années, intéressant les introns 7 et 8 du gène VCAN. Il en résulte une anomalie de l’épissage des exons 7 et 8, aboutissant à la production de protéines anormales et un déséquilibre du ratio des différents variants de la versicane (V0, V1, V2 et V4). Ceci est responsable d’une désorganisation microscopique du vitré avec sa liquéfaction très précoce, entraînant la formation d’une grande lacune caractéristique au fond d’oeil et une interface vitréorétinienne pathologique. À la lumière des dernières découvertes génétiques, une nouvelle classification émerge avec la description d’un groupe de pathologies liées aux mutations du chromosome 5q (vitréorétinopathies liées au gène VCAN), regroupant notamment le syndrome de Wagner, la vitréorétinopathie érosive et la rétinopathie de Jansen.
L’atteinte clinique est uniquement oculaire, antérieure et postérieure [9, 10]. Toute autre atteinte extra-oculaire doit faire reconsidérer le diagnostic. Du fait des similitudes avec la séméiologie oculaire du syndrome de Stickler, nous détaillerons plus loin ce tableau qui a longtemps servi de modèle de description des vitréorétinopathies héréditaires. Contrairement au syndrome de Stickler, l’atrophie choriorétinienne peut être sévère mimant parfois une choroïdérémie. Une atrophie optique est souvent présente dans les stades d’atrophie choriorétinienne avancés, traduisant la perte étendue des cellules ganglionnaires.
Il repose essentiellement sur le syndrome de Stickler qui associe des anomalies extra-oculaires (squelettiques, articulaires et ORL) à des lésions oculaires similaires, sans être identiques (myopie plus forte, vitré lacunaire avec condensation rétrolenticulaire, DR plus fréquents et précoces, atrophie choriorétinienne moins sévère).
Le pronostic visuel est surtout conditionné par l’évolution de l’atrophie choriorétinienne et la survenue d’un DR moins fréquent. Les patients jeunes sont plus à risque de présenter un DR rhegmatogène avec des déchirures souvent larges et multiples siégeant au niveau des cordons vitréens équatoriaux. Le risque de DR tractionnel augmente avec l’âge, lorsque les membranes prérétiniennes et adhésions vitréennes pathologiques se rétractent. Le pronostic postopératoire (cataracte, DR) est plus réservé que dans la population générale du fait de complications inflammatoires sévères fréquentes. Il semblerait en effet que la versicane soit impliquée dans des mécanismes de médiation de l’inflammation. Des cas d’uvéites sévères, sans autre étiologie retrouvée et souvent décompensés après une chirurgie oculaire, ont été rapportés dans deux familles.
Lorsqu’un cas de syndrome de Wagner est suspecté, il convient de réaliser un arbre généalogique exhaustif avec examen clinique des apparentés et un conseil génétique. Le traitement des complications rétiniennes et la problématique d’une attitude préventive sont les mêmes que dans le syndrome de Stickler et seront donc décrits ci-après.
P. Gascon, F. Matonti, P. Gastaud
Stickler a décrit, en 1965 puis 1967, un syndrome associant des anomalies vitréorétiniennes et des atteintes dégénératives squelettiques sous le nom d’arthro-ophtalmopathie progressive [11]. Ce syndrome affecte tous les groupes ethniques et son incidence est de l’ordre de 1 pour 10 000.
Ce syndrome est caractérisé par des mutations de gènes du collagène, pouvant entraîner une modification structurale des différents tissus contenant du collagène. Il s’agit d’une maladie autosomique dominante avec pénétrance quasi complète. Le Stickler de type 1 est associé à des mutations du gène COL2A1 codant pour le collagène de type II [12], alors que des mutations des gènes COL11A1 et COL11A2, codant pour le collagène de type XI, sont associées respectivement au Stickler de type 2 [13] et 3 [14]. Une formerécessive a été décrite dans certaines familles avec des mutations des gènes COL9A1 [15] et COL9A2 [16].
L’expression phénotypique est également dépendante de ces modifications génotypiques. Par exemple, le type 3 ne présente aucune atteinte oculaire, COL11A2 n’étant pas exprimé dans le vitré [17]. À l’inverse, il existe une forme oculaire pure du type 1 par une mutation limitée à l’exon 2 de COL2A1 [18]. L’atteinte vitréenne est également différente entre les types 1 et 2. Il est à noter que, du fait d’une expressivité variable, il peut exister de grandes différences phénotypiques au sein d’une même famille ou de patients non apparentés mais présentant la même mutation.
Dès 1987, il a été mis en évidence une relation entre le syndrome de Stickler et le gène codant pour le collagène de type II, qui est un homotrimère. À partir de 1996 et la classification de Snead, on sait que ce gène est associé au Stickler de type 1, ce qui n’est pas le cas du Stickler de type 2 associé à une mutation du gène du collagène de type XI [19–21].
Dans le Stickler de type 1, on retrouve plusieurs mutations différentes sans qu’il soit possible d’établir une corrélation entre le génotype et le phénotype [19, 22]. Sachant que le collagène de type II est le principal constituant (80 % ) des fibrilles de collagène vitréen et que les mutations dans le gène du collagène de type II affectent l’embryogenèse vitréenne, on retrouvera des anomalies congénitales n’évoluant pas au cours de la vie [18]. Dans le Stickler de type 2, cette liaison n’existe pas [23] et les seules mutations retrouvées concernent les gènes codant pour le collagène de type XI (hétérotrimère) [23, 24] qui est un constituant mineur des fibrilles vitréennes. On retient que dans ce cas-là, il y a un défaut de la fibrillogenèse tout au long de la vie expliquant l’aspect évolutif des modifications vitréennes du phénotype vitréen de type 2 [25]. Le Stickler de type 1 concerne près de 80 % des patients atteints de ce syndrome.
Des études histopathologiques et immunohistochimiques sur des prélèvements vitréens peropératoires ont montré la présence de cellules de Müller dans les cordages vitréens de la maladie [26–29]. Des cellules gliales modifiant l’interface vitréorétinienne pourraient intervenir dans la pathogénie des DR.
Le tableau clinique, variable en fonction des différentes mutations et de l’évolution, associe une atteinte oculaire (hormis pour le type 3), proche de celle de la maladie de Wagner, et l’apparition plus tardive de signes systémiques. Le diagnostic est essentiellement clinique. L’analyse génétique sera utile pour la confirmation du sous-type (Tableau 15-2), ainsi que pour proposer au patient et à sa famille un conseil génétique, tout en les informant de l’expressivité variable du syndrome.

Une myopie forte est présente dans 75 à 100 % des cas, importante d’emblée et peu évolutive ; la symptomatologie devient évidente principalement durant la deuxième décennie. On retrouvera : atteinte du segment antérieur, dégénérescence vitréorétinienne et déhiscences.
Un enfant sur trois développe une cataracte sous-capsulaire postérieure, le plus souvent bilatérale, avant l’âge de 20 ans. Une surveillance au long cours doit être effectuée, puisque 9 patients sur 10 auront un risque de développer une cataracte principalement nucléaire après 40 ans. Chez les enfants, on peut donc retrouver des opacifications du cristallin et plus particulièrement des opacités corticales en « quartier » bilatérales et symétriques (Fig. 15-13a). Initialement décrites par Seery, ces distinctive cataracts, présentes dans plus de 40 % des cas, pourraient constituer une méthode de dépistage efficace en cas de recherche systématique [30]. Des cas de glaucome chronique à angle ouvert ont aussi été rapportés.
Dans 85 à 100 % des cas, l’atteinte vitréenne est l’anomalie ophtalmologique la plus caractéristique. Les différents phénotypes vitréens, selon Richards et al., sont représentés dans la figure 15-14 : ils permettraient d’orienter l’analyse génétique de manière plus efficace qu’un dépistage à l’aveugle de toutes les mutations, selon le Tableau 15-2 [31]. Sur plus de 150 cas, l’efficacité de la détection mutationnelle après évaluation vitréorétinienne était positive dans 96,5 % des cas pour le phénotype membraneux/COL2A1 et dans 80 % des cas pour le phénotype perlé/COL11A1. L’aspect le plus typique est celui de membranes ou de rubans vitréens fenêtrés au milieu d’une cavité vitréenne optiquement vide (Fig. 15-15). On observe trois types de phénotypes vitréens, dont les plus fréquents sont les types 1 et 2 [19, 25] :
type 1 : secondaire à une mutation dans le gène du procollagène de type II (COL2A1), il correspond à 3 patients sur 4 [19] et se caractérise par un reliquat de vitré antérieur dans l’espace rétrolental (Fig. 15-13b) ; des membranes régulières s’insèrent au niveau de la pars plana (congenital membranous vitreous anomaly), plus rarement au niveau de l’équateur, et n’évoluent pas au cours de la vie puisqu’il s’agit de phénotypes vitréens congénitaux. Différentes mutations du gène du procollagène II ont été retrouvées dont une dans une position X (L467F) donnant un phénotype vitréen non fibrillaire (afibrillar congenital vitreous phenotype) [18, 25] ;
type 2 : en rapport avec une mutation sur le gène du collagène de type V/XI (COL11A1), le phénotype vitréen présente ici des anomalies de l’ensemble de la cavité vitréenne (Fig. 15-16) avec des cordages de diamètre variable associés à des zones localisées ou diffuses de décollement de la hyaloïde postérieure ; ces cordages sont décrits comme étant typiquement perlés (beaded congenital vitreous anomaly) ; il ne s’agit pas d’anomalies fixées mais évolutives. Des conversions phénotypiques du type 2 en type 1 ont été décrites, lorsqu’un décollement total de la hyaloïde postérieure se manifestant sous la forme d’une membrane vitréenne fenêtrée ramène le vitré en position antérieure ;
type 3 : à côté de ces deux types de phénotypes vitréens communs, un troisième a été identifié lorsque le vitré est moins dense, d’aspect hypoplasique par rapport à un vitré d’un sujet sain. Il a été associé à des mutations/phénotypes atypiques de COL2A1 [25], à des cas sporadiques dans des familles dont les individus étaient touchés par le phénotype membraneux [32], ainsi que chez des sujets avec des vitrés hypoplasiques [31]. Ce phénotype vitréen fait partie de la démarche diagnostique afin de rechercher la mutation causale.
Les anomalies rétiniennes retrouvées sont :
la dégénérescence palissadique : bilatérale, fréquente, très étendue et de topographie variable (Fig. 15-17) ;
des plages d’atrophie choriorétinienne et altérations pigmentaires : parfois observées dès la naissance, on les retrouve chez
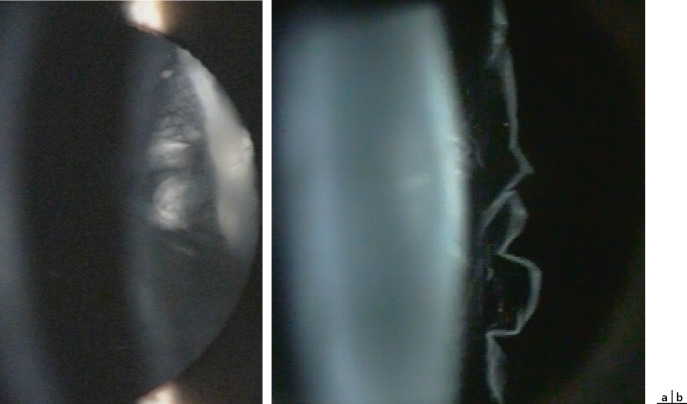
Fig. 15-13 Syndrome de Stickler type 1, segment antérieur.
a. Opacité cristallinienne en quartier. b. Aspect typique du vitré antérieur en « pseudo-collapsus vitréen » .
Fig. 15-14 Représentation schématique des phénotypes vitréens dans le syndrome de Stickler, d’après Richards et al. [31].
a. Corps vitréen normal, dense et homogène avec les lamelles de collagène dans la même direction. b. Phénotype vitréen membraneux situé dans l’espace rétrolental. c. Phénotype vitréen perlé avec lamelles irrégulières et épaissies. d. Phénotype vitréen hypoplasique moins dense, avec des lamelles irrégulières.
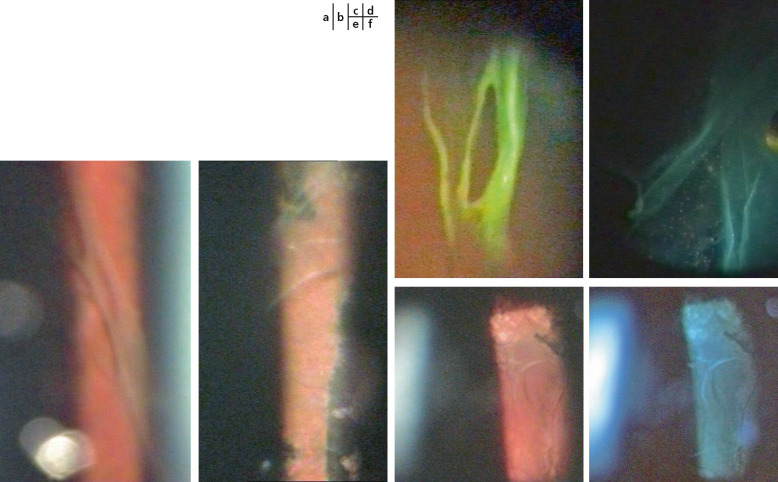
Fig. 15-15 Bandelettes ou rubans vitréens fenêtrés.
a. Rubans rétro-équatoriaux parallèles à l’ora. b. Rubans perpendiculaires à l’ora. c. Ruban fenêtré typique en lumière verte (photo peropératoire). d. Rubans vitréens tapissés de cellules inflammatoires. e, f. Rubans en arrière d’une photocoagulation circulaire équatoriale (mieux visible en lumière verte).
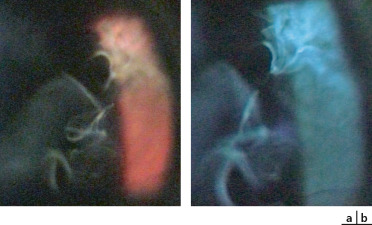
Fig. 15-16 Stickler type 2.
Les tractions vitréorétiniennes sont plus évidentes en lumière verte (b).

Fig. 15-17 Dégénérescence palissadique.
Les limites postérieures de la lésion apparaissent mieux en lumière verte (b).
2 patients sur 3 de manière circonférentielle en avant de l’équateur et/ou avec une topographie radiaire et périvasculaire en arrière de celui-ci. Dans le Stickler de forme oculaire pure, la dégénérescence rétinienne radiale périvasculaire serait une des clés du diagnostic clinique [33]. Quelques cas d’atrophies choriorétiniennes postérieures ont été aussi décrits [34].
Dans 75 % des cas, elles sont multiples, bilatérales et à des distances variables de l’ora serrata. Des trous atrophiques sont fréquemment retrouvés au niveau des palissades. Les zones de « blanc sans pression » sont des zones à risque puisque des déchirures géantes, à l’origine de 25 % des DR, surviennent volontiers chez le sujet jeune à ce niveau-là ; leur taux de bilatéralisation est voisin de 80 % .
Dans la majorité des cas, on retrouve des anomalies crâniofaciales associées aux atteintes ORL. Classiquement, ces patients présentent un faciès aplati avec un front large, un aplatissement de la base du nez, une petite lèvre supérieure et une micrognathie. Cet aspect caractéristique est plus marqué chez l’enfant et a tendance à disparaître avec l’âge. Aussi il est donc important lors de l’examen ophtalmologique d’un enfant, d’autant plus s’il présente une atteinte vitréorétinienne, de s’attarder sur l’examen global de la face [35]. Au niveau de la sphère ORL, on pourra retrouver :
une hypoacousie : présente dans plus de 80 % des cas, elle peut être soit de transmission soit neurosensorielle, soit une association des deux. Elle est très variable d’un individu à l’autre et différente entre les sous-types de Stickler (prédomine sur les hautes fréquences sonores dans le type 1, est plus diffuse dans le type 2 [36]). La surdité de transmission peut résulter d’une plus grande fréquence des otites séreuses moyennes (du fait de l’anatomie orale : fente palatine, palais ogival, etc.), d’une anomalie de la membrane tympanique ou d’une anomalie de mobilité de la chaîne des osselets. La perte auditive neurosensorielle est principalement associée à des mutations de COL11A1 et COL11A2, puisqu’elles modifient les propriétés mécaniques cochléaires ;
une fente palatine, isolée ou dans le cadre d’un syndrome de Pierre-Robin, des anomalies dentaires et un palais ogival.
Des luxations articulaires, en raison d’une hyperlaxité, peuvent se retrouver chez les patients plus jeunes. Puis avec l’âge, cette hypermobilité se réduit et la grande majorité des patients se plaignent de douleurs chroniques secondaires à des anomalies de la colonne vertébrale (scoliose, cyphose) ou à des malformations congénitales des hanches (coxa valga). Les autres articulations touchées sont les mains, les genoux et les chevilles. Ces différentes atteintes provoquent des séquelles arthrosiques au cours des troisième et quatrième décennies. Pouvant toucher jusqu’à 80 % des patients, elles peuvent avoir un retentissement considérable sur la qualité de vie.
Une association entre le syndrome de Stickler et le prolapsus de la valve mitrale a été retrouvée en proportion très variable selon les études.
Les complications ophtalmologiques sont dominées par le DR, conséquence des modifications architecturales vitréorétiniennes. Ainsi, 50 à 70 % des patients présenteront un DR, dont 25 % sur des déchirures géantes, et avec une grande proportion d’atteinte bilatérale. Le pronostic fonctionnel est plus sombre que dans la population générale du fait de l’ensemble des autres lésions associées (multiples déchirures, prolifération vitréorétinienne majeure) et du jeune âge des patients (entre 10 et 25 ans). De plus, lorsqu’un patient fait un DR sur l’oeil adelphe, son pronostic sera d’autant plus mauvais que le DR du premier oeil a été grave [37]. En fonction du mode évolutif, on distingue deux types de DR : les DR à progression lente compliquant des trous atrophiques et accompagnés de lignes de démarcation pigmentées ; les DR aigus tractionnels où la PVR est souvent présente d’emblée.
Du fait des anomalies visuelles et auditives, certains enfants pourront développer des troubles de l’apprentissage. Les enfants présentant des anomalies de la sphère buccale (fente palatine, syndrome de Pierre-Robin) nécessiteront une surveillance attentive du fait du risque vital respiratoire.
Une prise en charge multidisciplinaire (ophtalmologiste, ORL, pédiatre, généticien, rhumatologue, etc.) est nécessaire chez ces patients et leur famille. Devant le risque élevé de complications, une surveillance ophtalmologique régulière par un rétinologue est recommandée, dès le plus jeune âge. La fréquence des contrôles dépend du phénotype vitréen et également du contexte général, mais un examen tous les 6 à 12 mois est à prévoir.
Une des premières mesures de prévention est l’éducation du patient et de sa famille. Le pronostic visuel en cas de complication rhegmatogène est lié à la précocité de la prise en charge ; de ce fait, le patient et sa famille doivent connaître la conduite à tenir en urgence en cas de signes fonctionnels vitréorétiniens. De plus, il peut être recommandé de ne pas pratiquer certains sports traumatiques.
Devant la fréquence des DR, la question d’un traitement préventif interventionnel est essentielle chez ces patients et a fait l’objet de nombreux débats (Fig. 15-18). Peu d’études sur le sujet ont pu être réalisées et il s’agit uniquement d’études rétrospectives qui se sont principalement intéressées au type 1, du fait de son plus grand risque de DR. Une revue de la littérature de 2011 [38] a permis de retrouver un effet bénéfique d’un traitement préventif circonférentiel sur 360° soit par laser argon soit par cryothérapie, par rapport à un groupe contrôle non traité. Différents biais sont cependant notés dont le manque d’appariement pour l’âge. Malgré cela, il semble exister un bénéfice à pratiquer un traitement préventif.
Dans une étude rétrospective évaluant les résultats à long terme du protocole de cryothérapie prophylactique de Cambridge [39], 487 patients atteints de Stickler de type 1 ont été répartis, puis comparés, entre groupes traités uni- et bilatéralement versus groupes non traités uni- et bilatéralement. Les groupes ont été appariés par âge. Le groupe contrôle bilatéral avait 7,4 fois plus de risque de DR par rapport au groupe ayant bénéficié d’une cryothérapie prophylactique bilatérale sur 360° (p < 0,001). Le groupe contrôle unilatéral avait 10,3 fois plus de risque de DR par rapport au groupe ayant bénéficié d’une cryothérapie prophylactique unilatérale sur 360° (p < 0,001). Aucun cas d’effets secondaires graves n’a été rapporté avec ce traitement préventif (pas d’hémorragie choroïdienne, de plis maculaires ou de déficit visuel inexpliqué). Le principal but de cette technique est la prévention des décollements sur déchirures géantes.
Dans les formes graves, à haut risque, une indentation chirurgicale préventive a été proposée [37], malgré les difficultés d’indentation sur un oeil normotone (Fig. 15-19). Cependant elle ne peut être bénéfique que dans les cas où les tractions restent uniquement équatoriales, ce qui n’empêche pas malgré tout la survenue de déhiscences plus postérieures (Fig. 15-20) [9].
D’une manière plus globale, il s’agit avant tout de faire un bilan vitréorétinien complet afin d’évaluer les lésions, leur topographie ainsi que leur évolutivité. Des lésions rapidement évolutives ou un antécédent controlatéral nécessiteront plus volontiers un traitement prophylactique que des lésions stables peu étendues et/ou la présence de rubans vitréens proches de la macula pouvant faire opter pour une surveillance attentive [9].
Au total, il n’existe aujourd’hui aucun consensus sur une attitude préventive, celle-ci devant être décidée au cas par cas. La décision étant importante, il est parfois souhaitable qu’elle soit collégiale.
Dans le cadre de cette surveillance rétinienne, il est conseillé lors d’une chirurgie de la cataracte de choisir un implant de chambre postérieure à large optique permettant par la suite une visibilité optimale du segment postérieur.
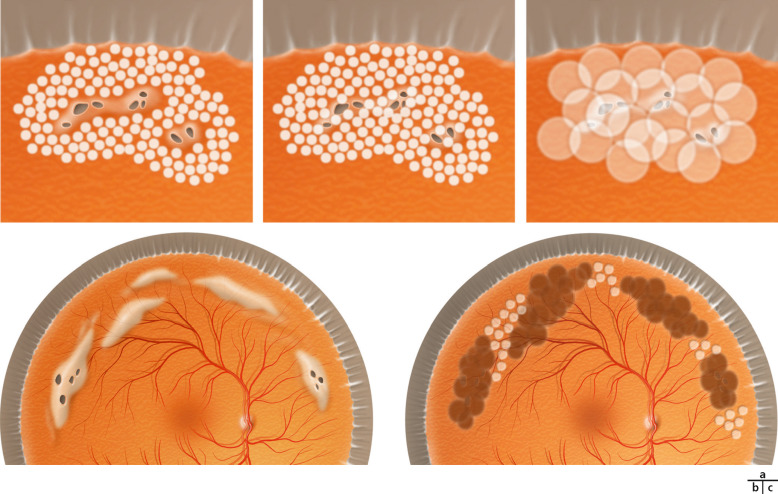
Fig. 15-18 Traitement préventif interventionnel.
a. Divers modes de rétinopexie préventive (photocoagulation entourant les lésions, couvrant les lésions et cryopexie) ; b. Lésions palissadiques circulaires. c. Traitement mixte : cryothérapie puis laser.
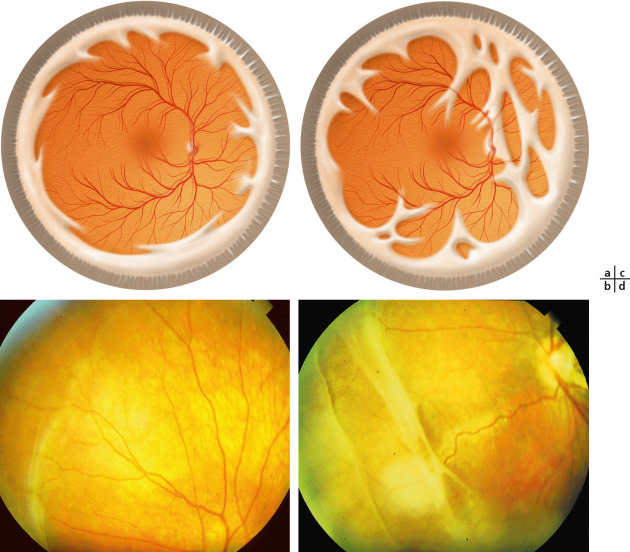
Fig. 15-19 Indentation chirurgicales préventive.
a, b. Tractions circonférentielles. c, d. Tractions circonférentielles et antéropostérieures, situation où une indentation circulaire est dangereuse.
(Source Fig. 15-19b et d : G. Brasseur, Pathologie du vitré, Rapport SFO, 2003.)
Fig. 15-20 Photocoagulations autour d’une déhiscence apparue en arrière d’une indentation circulaire.
En cas de DR rhegmatogène, la prolifération vitréorétinienne est présente dans près d’un cas sur trois et est fréquemment à l’origine des échecs chirurgicaux. La chirurgie endoculaire, bien que plus difficile à réaliser que dans la population générale, semble arriver à de meilleurs résultats que les autres techniques chirurgicales [40].
Une étude rétrospective récente a évalué les résultats après DR rhegmatogène, toutes causes confondues, traité en première intention par cryo-indentation sclérale sans prolifération vitréorétinienne significative initiale, chez des enfants de moins de 18 ans [41]. Si 73 % de l’ensemble des cas (n = 76/104) n’ont besoin que de cette seule chirurgie, les patients atteints de Stickler doivent dans 50 % des cas avoir au moins une autre chirurgie. Les procédures externes sembleraient donc moins efficaces, mais gardent leur place dans le traitement des DR non tractionnels.
Une prise en charge multidisciplinaire doit être réalisée chez ces patients. La période néonatale est à risque en cas de syndrome de Pierre-Robin puisqu’il peut y avoir une obstruction respiratoire par glossoptose. Les parents doivent donc recevoir une information sur le risque éventuel d’une position en décubitus dorsal chez ces enfants. L’alimentation doit également être surveillée attentivement devant les difficultés de succions et de déglutition et donc la possibilité de faire des fausses routes. À partir de l’âge de 4 mois, l’alimentation sera plus aisée, car ils pourront commencer à s’alimenter à la cuillère. Toutes les autres anomalies peuvent bénéficier d’une prise en charge spécialisée pour les patients et leur famille.
On distingue deux catégories de diagnostics différentiels : les atteintes syndromiques et celles non syndromiques. On retiendra principalement, dans la première catégorie, le syndrome de Marshall sans arthropathie. Dans les atteintes vitréorétiniennes isolées, il faudra évoquer le syndrome de Wagner beaucoup moins fréquent, mais dont les manifestations sont extrêmement proches, avec en plus une héméralopie et une myopie plus modérée. Les autres diagnostics à évoquer sont les autres hérédodégénérescences vitréorétiniennes. Il existe par ailleurs des cas isolés de dystrophie vitréorétinienne chez des sujets très myopes, avec une séméiologie très proche et posant les mêmes problèmes de prévention et de traitement : ces syndromes « Stickler-like » sont parfois en rapport avec des séquelles mineures de rétinopathie des prématurés.
Le syndrome de Stickler est donc une pathologie oculaire grave à cause de la fréquence et de la gravité du DR. Un dépistage doit être organisé le plus précocement possible afin de proposer l’attitude préventive la plus adaptée au patient. Un suivi fréquent et régulier au long cours est ainsi recommandé.
F. Metge-Galatoire
Le rétinoschisis juvénile lié à l’X (RSJ) est une maladie rétinienne congénitale bilatérale. Elle se caractérise par la présence d’un clivage anormal de la rétine centrale et périphérique, responsable d’une baisse visuelle progressive et de complications à type d’hémorragie intravitréenne et de DR. Décrit pour la première fois par Haas en 1898 [42], il est la cause la plus fréquente de dégénérescence maculaire juvénile chez le garçon. Sa prévalence est estimée de façon assez variable selon la localisation géographique entre 1/5000 à 1/25 000 [43]. Son mode de transmission est récessif lié à l’X, avec une pénétrance complète et une expressivité variable.
Le diagnostic de la maladie est souvent posé chez le garçon d’âge scolaire ou préscolaire, typiquement entre 5 et 10 ans, devant des difficultés à la lecture. C’est plus rarement un strabisme qui révèle l’affection dans la petite enfance. L’atteinte fonctionnelle est bilatérale mais le plus souvent nettement asymétrique [44]. Une forte hypermétropie axile est souvent associée au RSJ. Il existe une très grande hétérogénéité anatomique et fonctionnelle d’un individu à l’autre, mais également au sein d’une même famille, avec un retentissement fonctionnel plus ou moins précoce et plus ou moins important pour une même mutation.
L’acuité visuelle se détériore habituellement de façon significative pendant les deux premières décennies pour se situer aux alentours de 2 à 5/10, puis reste souvent relativement stable jusqu’à la cinquième ou sixième décennie. L’apparition secondaire d’une atrophie maculaire lentement évolutive peut s’accompagner d’une nouvelle aggravation de la baisse visuelle au-delà de 60 ans [43].
Sur le plan anatomique, le RSJ se caractérise typiquement par un aspect maculaire microkystique stellaire bilatéral, centré sur la fovéola, avec un aspect en « rayon de roue » ou en « pétale de fleur » (Fig. 15-21) qui est retrouvé dans 95 à 100 % des cas [45]. Cet aspect a pu être décrit chez le très jeune enfant (< 3 mois) [46], cependant il peut être absent à un stade précoce et apparaître au cours de l’évolution de la maladie pour concerner, tôt ou tard, 100 % des patients. L’aspect typique de la macula se modifie avec le temps et prend des aspects multiples, les cavités kystiques ayant tendance à progressivement coalescer. Après 50 ans, des remaniements pigmentaires associés à un certain degré d’atrophie maculaire sont fréquents ; l’évolution ultime se fait vers l’atrophie maculaire plus ou moins étendue. L’atteinte
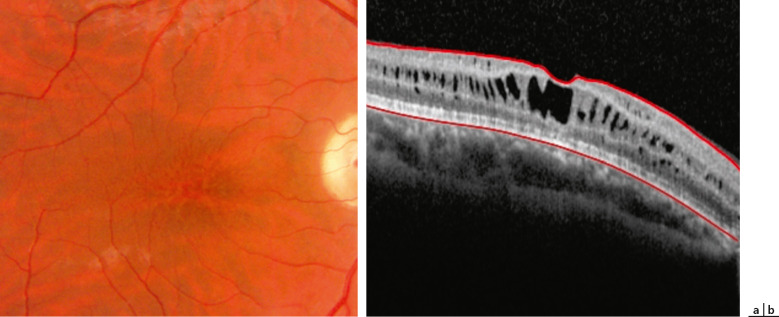
Fig. 15-21 Aspect maculaire en rayon de roue.
En OCT (b), clivage intrarétinien prédominant au niveau de la plexiforme interne.
anatomique, comme l’atteinte fonctionnelle, peut être très asymétrique chez un même individu [47].
Un rétinoschisis périphérique unique ou multiple est associé à l’atteinte maculaire dans 40 à 50 % des cas, plus souvent localisé en temporal inférieur [43] ; il est initialement bulleux et transparent. Son évolution peut se faire, avant l’âge de 10 ans le plus souvent, vers la résolution spontanée, laissant place à des plages de pigmentations ou de dépigmentations périphériques, ou vers l’apparition de déhiscences du feuillet interne parfois très étendues et périvasculaires, laissant certains vaisseaux dépourvus de support rétinien et flottants dans la cavité vitréenne (Fig. 15-22). La survenue d’une hémorragie intravitréenne est possible au cours de l’évolution et peut être le mode de révélation de la maladie, au point que toute hémorragie intravitréenne unilatérale chez le garçon occasionnée par un traumatisme, minime ou a fortiori spontané, doit faire évoquer le diagnostic ; le DR, plus rare, est exceptionnellement inaugural.
Le rétinoschisis périphérique est pourvoyeur des deux principales complications du RSJ qui sont l’hémorragie intravitréenne et le DR (Fig. 15-23), complications plus fréquentes dans les deux premières décennies de la vie.
L’hémorragie intravitréenne et/ou intrakystique observée dans 4 à 40 % des cas est due à la rupture d’un vaisseau en pont ou plus rarement à une néovascularisation secondaire. Lorsque l’hémorragie est inaugurale, c’est la tomographie cohérence optique (optical coherence tomography [OCT]) du deuxième oeil qui orientera le diagnostic en montrant les anomalies maculaires caractéristiques du rétinoschisis.
Le DR complique le RSJ dans 5 à 22 % des cas ; il est lié à la présence concomitante de déhiscences dans les feuillets interne et externe de la rétine. Les déhiscences du feuillet externe sont beaucoup plus rares que dans le feuillet interne, ce qui explique que le DR soit relativement peu fréquent [43].
Les clichés en autofluorescence montrent très clairement l’aspect microkystique radiaire en rayon de roue isofluorescent de la macula.
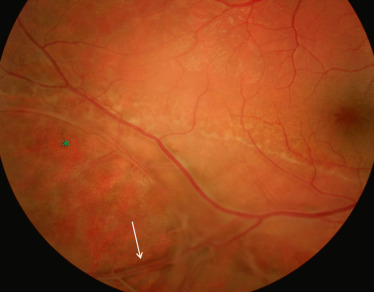
Fig. 15-22 Présence d’un rétinoschisis périphérique chez un enfant de 12 ans.
L’astérisque (*) montre une déhiscence dans le feuillet interne de la rétine. Un vaisseau flottant est visible au sein de la vaste déhiscence inférieure (flèche).
L’OCT est aujourd’hui l’examen de premier plan pour le diagnostic de la maladie ; en mode « en face » l’aspect en rayon de roue est particulièrement démonstratif (Fig. 15-24). En coupes, l’OCT objective le clivage intrarétinien et permet de mieux analyser l’évolution de la maculopathie au cours du temps. L’image caractéristique est celle d’un clivage intrarétinien d’importance et de localisation variable (Fig. 15-21, 15-25 et 15-26).
Dans une analyse détaillée récente de l’atteinte maculaire en OCT, portant sur 20 yeux de patients atteints de RSJ, âgés en moyenne 17,6 ans et comparés à un groupe témoin, Yang et al. [48] retrouvent la présence d’un fovéoschisis dans 85 % des cas et une atteinte périphérique dans 55 % des cas ; 15 % des patients présentent une atteinte périphérique isolée et 45 % une atteinte fovéolaire isolée. La présence de défects dans la couche des photorécepteurs est observée dans 75 % des cas ; le rétinoschisis intéresse de façon égale la rétine interne dans 85 % des cas et la rétine externe (85 % des cas). Au niveau de la rétine externe, les structures préférentiellement intéressées par le rétinoschisis sont la plexiforme externe (60 % des cas) et la ligne correspondant à l’extrémité des articles externes des cônes (cone outer segment tip line [COST line]) qui apparaît irrégulière dans 75 % des cas et associée à un raccourcissement des articles externes des photorecepteurs.
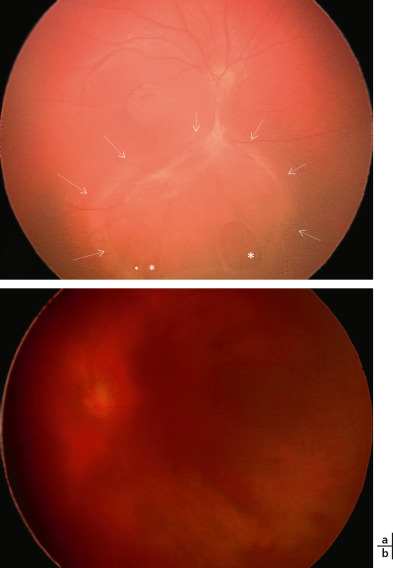
Fig. 15-23 Enfant présentant une hémorragie intravitréenne de l’oeil gauche (b) et un rétinoschisis visible au niveau de l’oeil droit (a).
La macula paraît cliniquement partiellement atrophique. Le rétinoschisis est inférieur avec de petites déhiscences dans le feuillet interne (*) et une vaste déhiscence dans le feuillet externe (flèches).
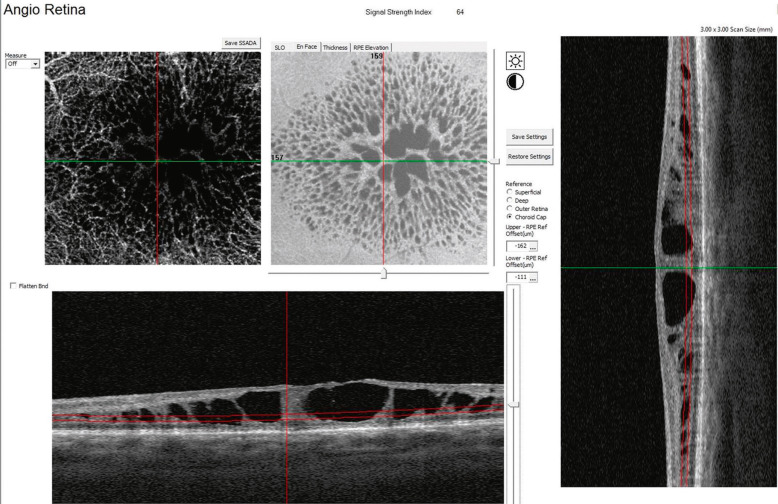
Fig. 15-24 En OCT « en face » , noter l’aspect en rayon de roue et la correspondance en OCT bidimensionnel.
(Remerciements au Dr B. Haouchine.)
La mauvaise acuité visuelle paraît nettement corrélée à l’importance des anomalies de la rétine externe (défects dans la COST line et raccourcissement des segments externes des photorécepteurs).
Au cours du temps, la maculopathie peut évoluer vers la constitution d’un kyste maculaire unique (Fig. 15-27) par coalescence progressive des cavités schisiques ; des aspects de trou maculaire ou de trou lamellaire ont également pu être observés [49]. Enfin aux stades tardifs de la maladie apparaissent des plages d’atrophie avec disparition du clivage rétinien et amincissement de la rétine (Fig. 15-28).
L’OCT est également utile pour différencier rétinoschisis et DR ou faire le diagnostic de DR associé au rétinoschisis périphérique, en particulier lorsque le rétinoschisis s’entend de façon assez postérieure, l’analyse de la rétine périphérique étant plus difficile en OCT (Fig. 15-29).
L’angiographie à la fluorescéine peut avoir un intérêt en cas de doute avec un oedème maculaire cystoïde. Aucune hyperfluorescence maculaire n’est notée en angiographie en cas de rétinoschisis en l’absence d’atrophie associée.
L’électrorétinogramme (ERG) était un examen incontournable dans le diagnostic de la maladie avant l’avènement de l’OCT, il garde aujourd’hui un intérêt dans les formes atypiques en particulier lorsque le profil maculaire est conservé en OCT sur l’un des deux yeux, ou dans les formes évoluées de la maladie compliquées d’atrophie maculaire. L’ERG montre typiquement une réduction de l’amplitude de l’onde b et une relative préservation de l’onde a avec un rapport b/a diminué. Avec l’âge et dans les formes associées à une atrophie étendue de l’épithélium pigmentaire, l’onde a peut, elle aussi, être diminuée. Dans de rares cas, l’onde b est peu diminuée en ERG global, mais est généralement diminuée en ERG multifocal maculaire. Les modifications de l’ERG avec diminution de l’onde b ont initialement étayé l’hypothèse d’une anomalie des cellules de Müller dans le déterminisme de la maladie, d’autant plus que l’analyse histologique de la rétine de patients décédés montrait l’accumulation de matériel filamenteux apparemment issu des cellules de Müller. L’identification du gène responsable de la maladie en 1997 a permis de montrer l’implication des photorécepteurs et des cellules bipolaires et non des cellules de Müller dans le processus pathologique [50].
Le mode de transmission du RSJ, mis en évidence en 1913, est récessif lié à l’X avec une pénétrance complète et une expressivité variable. La maladie est due à des mutations du gène RS1 composé de 6 exons et localisé sur le bras court du chromosome X en position Xp22.13. Ce gène code la rétinoschisine (protéine constituée de 224 acides aminés, sécrétée par les photorécepteurs et les cellules bipolaires) qui joue un rôle crucial dans les interactions et les adhérences intercellulaires dans la rétine, en particulier l’adhérence synaptique entre les photorécepteurs et les cellules bipolaires, et possiblement un rôle dans les mouvements de fluides entre les secteurs intra- et extracellulaires [50]. En immunomarquage, la rétinoschisine est retrouvée au niveau des segments internes des photorécepteurs, des cellules bipolaires, et au niveau des plexiformes internes et externes de la rétine ; elle n’est pas retrouvée au niveau des cellules de Müller. À ce jour, plus de 200 mutations différentes du gène RS1 ont été répertoriées comme associées au rétinoschisis.
Ces mutations sont majoritairement de type faux sens et dans ce cas concernent préférentiellement les exons 4, 5 et 6 du gène qui constituent le domaine discoïdine. Les autres types de mutations (nonsens, délétions, insertions, mutations introniques) intéressent toutes les régions de la protéine RS1 de façon plus homogène. Aucune corré-
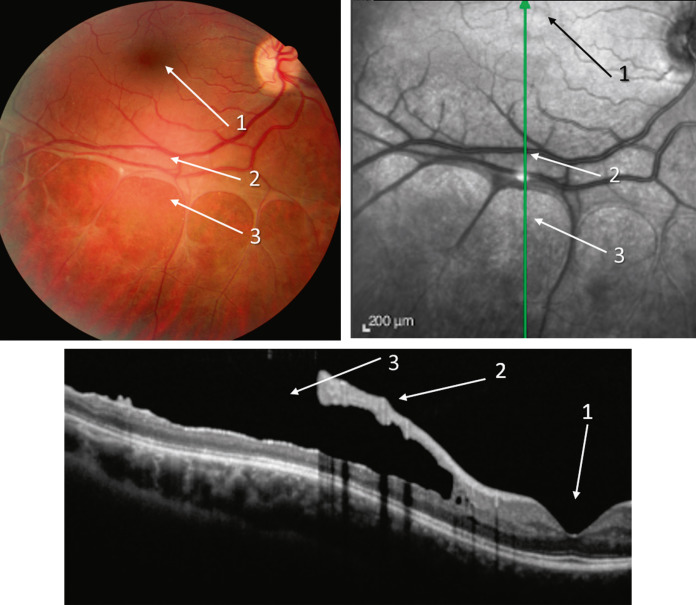
Fig. 15-25 Rétinoschisis juvénile (RSJ) chez un patient dont l’acuité visuelle est de 10/10.
Noter l’aspect maculaire normal (1), le rétinoschisis périphérique (2) et les déhiscences dans le feuillet interne de la rétine (3).
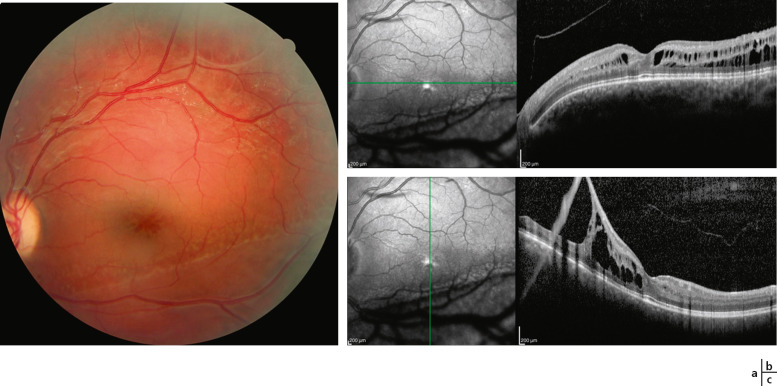
Fig. 15-26 Rétinoschisis juvénile (RSJ) lié à l’X.
a. Aspect maculaire caractéristique en pétale de fleur sur le cliché en couleurs. b, c. En coupe horizontale (b), la dépression fovéolaire est conservée, mais en coupe verticale (c), le rétinoschisis péripherique s’étend jusqu’à à la partie inférieure de la macula (acuité visuelle 8/10).
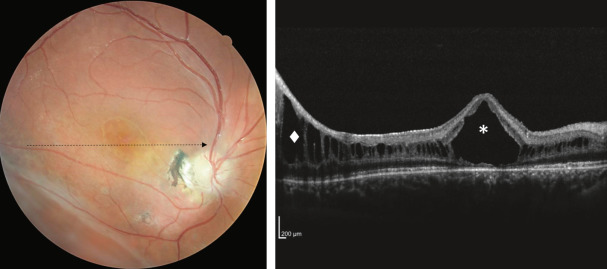
Fig. 15-27 Vaste kyste centromaculaire (* en a) chez un enfant de 11 ans avec atrophie maculaire en arrière du kyste et présence d’un rétinoschisis périphérique (♦ en b) (acuité visuelle 1/10).
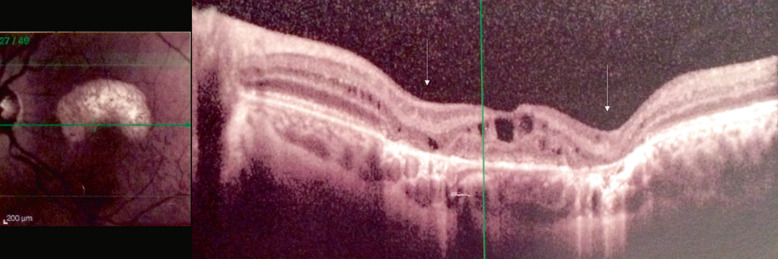
Fig. 15-28 Atrophie supéromaculaire épargnant le centre de la macula chez un jeune homme de 17 ans (acuité visuelle 5/10).
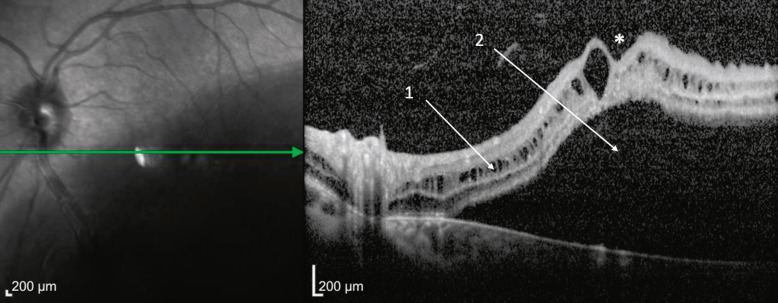
Fig. 15-29 Jeune homme de 18 ans atteint de RSJ (1) compliqué de décollement de rétine (2). La macula est décollée (*).
lation significative n’a été retrouvée entre génotype et phénotype à ce jour en dehors d’une étude de Vincent et al. [51] qui observe des anomalies moins marquées de l’ERG en cas de mutation faux sens.
Récemment, des essais de thérapie génique chez la souris rendue déficiente en rétinoschisine ont donné des résultats encourageants puisqu’une restauration significative de la structure et de la fonction rétinienne a pu être obtenue [50, 52]. Deux études de thérapie génique de phase I/II sont actuellement en cours chez l’homme aux États-Unis [53, 54].
Le diagnostic de rétinoschisis chez un jeune garçon implique un bilan ophtalmologique complet avec réfraction sous cycloplégie de façon à dépister l’hypermétropie fréquemment associée à la maladie et prescrire la correction optimale. Si une amblyopie relative existe, elle a souvent une double part, anatomique et fonctionnelle, en particulier en cas de strabisme associé ; une rééducation est alors nécessaire pour gagner sur la part fonctionnelle avant l’âge de 10 ans.
Des photos du fond d’oeil et une OCT seront systématiques pour faire un état des lieux de la maladie et permettre l’évaluation objective de son l’évolutivité au cours du temps. Un ERG est habituellement réalisé même si dans les cas typiques, il n’est pas impératif pour établir le diagnostic.
En l’absence de complications, aucun traitement n’est habituellement proposé, en particulier aucun traitement laser ni au bord d’un rétinoschisis périphérique ni en son sein. Le laser n’empêche en aucune manière la progression du rétinoschisis et ne présente que très rarement un intérêt pour traiter une éventuelle déhiscence du feuillet externe au sein d’un rétinoschisis périphérique. Plusieurs cas de DR secondaires à une photocoagulation ont été décrits dans la littérature, en raison de la survenue de déhiscences iatrogènes sur le feuillet externe du rétinoschisis [43]. Il faudra déconseiller la pratique de sports de combat et de sports violents pour minimiser le risque d’hémorragie intravitréenne.
Le traitement préventif des complications du RSJ reste aujourd’hui débattu, Yu [55] propose la réalisation d’une vitrectomie systématique pour freiner l’évolution de la maculopathie et prévenir la survenue de complications dans une étude prospective non randomisée publiée en 2013. Sur une série de 17 yeux d’enfants âgés en moyenne de 12 ans, il évalue l’efficacité de la vitrectomie (avec décollement du vitré peropératoire, pelage de la limitante interne et tamponnement par gaz) par rapport à un groupe témoin (11 yeux) avec un recul moyen de 34,7 mois (10 à 63 mois). Cet auteur retrouve, de façon statistiquement significative, une amélioration de l’acuité visuelle dans le groupe traité de même qu’une amélioration du profil maculaire en OCT avec disparition du rétinoschisis maculaire et un taux de complications secondaires moindre après chirurgie (DR rhegmatogène de 28 % dans le groupe témoin versus 12 % dans le groupe traité). Mais ces résultats restent isolés, car les publications sur le sujet sont rares et aucun consensus n’existe sur la prise en charge des rétinoschisis avec maculopathie évolutive en l’absence de complication. La surveillance simple reste l’attitude la plus répandue.
Cette complication survient le plus souvent dans les deux premières décennies de la vie, alors que le vitré n’est pas décollé ; dans la majorité des cas, la vitrectomie n’est pas nécessaire et la résorption du sang se fait sans séquelle. Une vitrectomie est indiquée si l’hémorragie est plusieurs fois récidivante ou très dense sans tendance à la résorption rapide. La vitrectomie permettra la coagulation du ou des vaisseaux responsables. Lorsque le vitré n’est pas dissécable, une ablation plus ou moins complète du feuillet interne de la rétine est parfois nécessaire pour supprimer les tractions du vitré sur la rétine [56].
Il est secondaire à une déhiscence des feuillets interne et externe de la rétine au sein du schisis ou en rétine saine. Au sein du schisis périphérique, les trous du feuillet interne sont facilement repérables mais ne sont pas seuls à l’origine du DR. En revanche, la recherche des trous au niveau du feuillet externe peut s’avérer plus difficile : ils se situent le plus souvent au niveau du bord postérieur du schisis périphérique, leur traitement est indispensable à la réapplication de la rétine. Une chirurgie ab externo peut être réalisée dans ce type de DR, en l’absence de déchirure trop postérieure ou de prolifération vitréorétinienne, mais il existe un risque élevé de prolifération vitréorétinienne dans les suites avec un taux de récidive de décollement proche de 40 % [57]. Un abord endoculaire est donc souvent nécessaire.
Dans cette pathologie, les DR tractionnels s’étendent postérieurement au rétinoschisis périphérique et sont probablement favorisés par une très grande adhérence entre le cortex vitréen et le feuillet interne du rétinoschisis périphérique. Pour ces DR, une chirurgie endoculaire est réalisée, à type de vitrectomie avec décollement de la hyaloïde postérieure si possible ; une rétinectomie du mur interne du schisis est faite lorsque la hyaloïde n’est pas dissécable, ainsi qu’une cautérisation des vaisseaux flottants associée à un tamponnement interne par gaz ou silicone [58].
Le résultat anatomique et fonctionnel de la chirurgie du DR compliquant un RSJ est plutôt encourageant, avec un taux de réapplication proche de 85 % [59, 60] et une amélioration de l’acuité visuelle dans 50 à 67 % des cas. Compte tenu de la rareté de cette maladie, les études publiées à ce jour sur les DR sur rétinoschisis sont très peu nombreuses et ont un très faible effectif ; les résultats doivent donc être interprétés avec réserve.
Une enquête génétique doit être proposée systématiquement à tous les sujets atteints, ainsi qu’aux possibles conducteurs et à tous les membres de la famille en général. Le mode de transmission lié à l’X sera expliqué. En dehors d’un mariage consanguin, dans la descendance d’un homme atteint de rétinoschisis, toutes les filles seront obligatoirement conductrices et aucun des fils ne sera atteint ; dans la descendance d’une femme conductrice le risque d’avoir un fils atteint sera de 50 % de même que celui d’avoir une fille conductrice (50 % ). Du fait de la transmission avec pénétrance complète, tous les hommes porteurs de la mutation seront atteints mais de façon très variable au sein d’une même famille en raison de l’expressivité variable de la mutation.
Cas particulier des femmes conductrices : le RSJ lié à l’X peut atteindre les femmes, dans de rares cas de familles consanguines, et dans le cas encore plus rare d’association de la mutation à un syndrome de Turner (45/X). En dehors de ces circonstances exceptionnelles, les femmes conductrices sont saines et aucune anomalie ophtalmologique attribuable à la mutation n’est habituellement détectable, sur le plan anatomique et fonctionnel. Il semble exister un consensus sur le fait que les quelques anomalies rapportées dans la littérature chez des femmes conductrices soient non spécifiques et non caractéristiques de la mutation (palissades, membranes épimaculaires, rétinoschisis périphérique d’allure sénile chez des femmes d’âge mûr, etc.) [43].
Le RSJ est la dégénérescence maculaire juvénile la plus fréquente chez le garçon et s’accompagne d’une maculopathie d’aspect caractéristique en OCT et en ERG. Il existe une très grande hétérogénéité clinique y compris dans une même famille pour une même mutation, mais les formes sévères, compliquées d’hémorragie intravitéenne ou de DR s’observent essentiellement chez le jeune enfant. L’évolution de la maladie est lente et, le plus souvent, compatible avec une acuité visuelle longtemps utile sur au moins l’un des deux yeux. L’apparition d’une atrophie maculaire très invalidante survient en général après l’âge de 60 ans. Des espoirs de thérapie génique existent aujourd’hui et font l’objet d’études cliniques chez l’homme.
C. L andré, P. Gastaud
La vitréorétinopathie exsudative familiale (VREF) a été décrite pour la première fois en 1969 par Criswick et Schepens [61]. Il s’agit d’une affection bilatérale et progressive, liée à une anomalie de développement de la vascularisation rétinienne périphérique créant une zone avasculaire antérieure qui entraîne, du fait de l’ischémie, des complications néovasculaires proches de ce que l’on peut rencontrer dans la rétinopathie du prématuré.
Pour décrire la VREF, on peut s’appuyer sur la classification de Pendergast et Trese datant de 1998 et réalisée à partir de l’observation de 52 yeux de 26 patients. Le stade 1 est caractérisé par une zone périphérique avasculaire, le plus souvent située en temporal. Le stade 2 regroupe les patients présentant une néovascularisation associée à cette zone avasculaire. Ce groupe est divisé en deux sous-groupes : le groupe 2a sans signe d’exsudation et le groupe 2b accompagné de phénomènes exsudatifs. Le stade 3 correspond à la présence d’un DR sub-total épargnant la macula (stade 3a en cas de décollement exsudatif et stade 3b en cas de décollement tractionnel). Le stade 4 est caractérisé par un DR subtotal incluant la macula (stade 4a si le décollement est exsudatif et stade 4b si le décollement est tractionnel). Enfin, le stade 5 correspond à un DR total (stade 5a avec tunnel ouvert et stade 5b avec tunnel fermé) [61].
En parallèle de cette description, d’autres signes sont aussi assez caractéristiques de la VREF. Il s’agit principalement des zones de « blanc sans pression » et du pli radiaire temporal correspondant à une traction et un plissement de la macula, associé à un étirement horizontal des vaisseaux. On retrouve en outre des hémorragies rétiniennes et des zones d’atrophie choriorétinienne [62].
La VREF est une pathologie dont la présentation clinique est fréquemment asymétrique et dont l’expression est grandement variable chez les différents individus d’une même famille. Par ailleurs, elle est plus fréquente en Asie que dans le reste du monde.
Dans la grande majorité des cas, la VREF est transmise de manière autosomique dominante. Cependant, il existe des formes de transmissions liées à l’X, autosomiques récessives ou de novo.
Il existe six principaux gènes connus dont la mutation provoque la VREF. Il s’agit de FZD4, LRP5, TSPAN12, NDP et ZNF408 et KIF11. Une mutation d’un de ces gènes est retrouvée dans environ 50 % des cas de VREF. Quatre de ces gènes sont impliqués dans la voie de signalisation Wnt qui semble jouer un rôle capital dans la vascularisation rétinienne.
FZD4, situé dans la région 11q13-q23, est en cause principalement dans les formes autosomiques dominantes. LRP5, qui est principalement responsable de formes autosomiques récessives, se trouve dans une région proche de FZD4. Sa mutation peut aussi entraîner un syndrome d’ostéoporose-pseudogliome. TSPAN12 se situe au niveau du chromosome 7q31.31. Sa mutation a été montrée de manière homozygote chez des familles atteintes de VREF selon un mode autosomique récessif. NDP est situé au niveau du chromosome Xp11.4 : il est donc responsable de formes avec transmission liée à l’X. Il est aussi impliqué dans la maladie de Norrie. ZNF408 est situé sur le chromosome 11p11.2 et est responsable de formes autosomiques dominantes. KIF11, situé sur le chromosome 10q24.1, est impliqué dans des formes à transmission autosomique dominante. Sa mutation a d’abord été mise en évidence dans un spectre de pathologies associant microcéphalies, lymphoedème, retard mental et dysplasie choriorétinienne non identifiée avant d’être mis en évidence chez des patients atteints de VREF.
Un traitement est nécessaire devant une exsudation ou une néovascularisation et a fortiori en présence d’un DR. La première ligne thérapeutique est constituée par la photocoagulation au laser argon. Pendergast et al. ont montré que la photocoagulation effectuée devant une exsudation sous-rétinienne ou une néovascularisation extrarétinienne avait une bonne efficacité avec un maximum de trois séances ; la grande majorité des yeux classés en stade 2b et ainsi traités ne nécessitait pas de vitrectomie par la suite. Le traitement par laser peut aussi être proposé pour les yeux présentant un DR localisé, afin de réduire la néovascularisation et les exsudations avant la vitrectomie [61].
La cryothérapie est proposée par certains auteurs en prévention du DR en cas d’échec de la photocoagulation, mais son utilisation n’est pas consensuelle [63].
En cas de DR, un traitement par vitrectomie et/ou indentation peut être proposé [64].
Plus récemment, l’utilisation de bévacizumab a été essayée dans la prise en charge des néovascularisations de la VREF avec des résultats encourageants.
Le pronostic de la VREF dépend de la précocité d’apparition des lésions dans l’enfance. Il est logiquement plus sombre en cas de complications à un âge jeune. Par ailleurs, il est admis que les formes asymétriques sont de meilleur pronostic car souvent l’un des deux yeux conserve une acuité visuelle fonctionnelle (Fig. 15-30). D’une manière générale, le pronostic est sombre avec une cécité légale pouvant atteindre 20 % des cas [65].
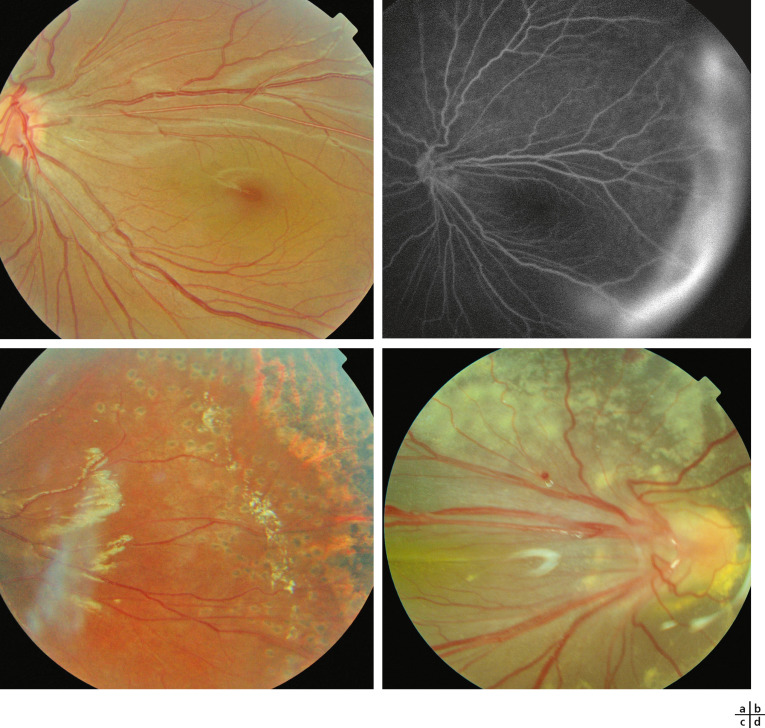
Fig. 15-30 Vitréorétinopathie exsudative familiale (VREF).
a, b. VREF stade 2a oeil gauche avant traitement. c. Néovaisseaux déshabités après cryothérapie. d. OEil droit stade 4b : DR persistant malgré le tamponnement par huile de silicone.
Le principal diagnostic différentiel de la VREF est la rétinopathie du prématuré. Les éléments cliniques permettant de les différencier sont principalement l’absence de prématurité ou d’hypotrophie chez les enfants atteints de VREF, ainsi que l’histoire familiale. Les autres diagnostics différentiels principaux sont la maladie de Coats, la persistance de la vascularisation foetale, le rétinoblastome et l’incontinentia pigmenti [62].
La maladie de Norrie et l’ostéoporose-pseudogliome présentent des mutations génétiques communes à la VREF et sont donc à la limite entre diagnostics différentiels et présentations différentes d’une même maladie.
C. Landré
La vitréorétinopathie érosive a été décrite pour la première fois en tant que nouvelle entité par Brown et al. en 1994 [66]. Elle est décrite comme associant une dystrophie vitréenne à des anomalies de l’épithalium pigmentaire rétinien.
Les enfants atteints présentent progressivement des anomalies du champ visuel associées à une héméralopie. À l’examen du segment postérieur, on retrouve des anomalies vitréennes à type de zones optiquement vides (appelées vitreous syneresis) et constituées de vitré liquéfié. Par ailleurs, il existe des anomalies de l’épithélium pigmentaire à type d’érosions avec visualisation anormale de la vascularisation choroïdienne : ces « érosions épithéliales » débutent plutôt en périphérie, mais peuvent conduire à une atrophie postérieure majeure chez les patients plus âgés. Enfin, ces anomalies se compliquent fréquemment de DR tractionnels et/ou rhegmatogènes. À l’électrocardiogramme (EEG), on observe des anomalies de type rod/cone [66]. On ne retrouve dans cette pathologie ni myopie, ni anomalies orofaciales ou systémiques.
Une mutation génétique associée à la vitréoretinopathie érosive a été retrouvée sur le chromosome 5q14.3, il s’agit de la mutation CSPG2/Versican qui est aussi en cause dans le syndrome de Wagner [67, 68]. La transmission se fait probablement sur un mode autosomique dominant.
La prise en charge thérapeutique est dominée par le traitement du DR. Les caractéristiques anormales du vitré rendent la chirurgie plus compliquée et d’issue plus incertaine.
Le principal diagnostic différentiel est la maladie de Wagner qui présente aussi des anomalies vitréennes et de l’épithélium pigmentaire. Pour certains auteurs, on ne peut pas réellement parler de diagnostic différentiel mais plutôt d’expression variable de la même pathologie. Les autres diagnostics différentiels sont principalement le syndrome de Stickler et le syndrome de Goldmann-Favre qui présentent aussi des anomalies vitréennes similaires, mais des anomalies différentes de l’épithélium pigmentaire et des anomalies générales, notamment de dysmorphies faciales associées.
Le pronostic des patients atteints de vitréorétinopathie érosive reste sombre. Un DR apparaît dans environ 50 % des cas durant l’évolution. Par ailleurs, l’érosion de l’épithélium pigmentaire qui commence en périphérie altère notablement le champ visuel et peut conduire à une cécité par extension au pôle postérieur.
B. Butet, P. Gastaud
Il s’agit d’une vitréorétinopathie héréditaire rare caractérisée par une dégénérescence vitréenne associée à des éléments atypiques de rétinopathie pigmentaire. Les premiers éléments syndromiques ont été décrits par Goldmann puis Favre [69] en 1958 chez une fratrie atteinte d’héméralopie précoce. Les patients atteints de ce syndrome présentent notamment une hyperexpression et sensibilité des cônes S ( « bleus » ) ; il s’agirait d’une forme parente du syndrome d’augmentation des cônes bleus (enhanced S cone sensitivity syndrome).
C’est une des vitréorétinopathies héréditaires les plus rares, la prévalence est estimée à moins d’un cas pour un million [70]. Il ne semble pas y avoir de prédominance homme-femme.
La transmission est autosomique récessive. La maladie est causée par différentes mutations du gène NR2E3 (localisé sur le chromosome 15q23 ; anciennement PNR pour photoreceptor-specific nuclear receptor) impliqué dans le développement et la différenciation des photorécepteurs au cours du développement. Plus de 40 mutations différentes ont déjà été répertoriées. Une altération de ce gène aboutit à la dégénérescence des bâtonnets et un ratio anormal des différents cônes, avec une hyperprésence des cônes S (short ou bleus) au détriment des cônes M (medium ou verts) et L (long ou rouges) [71]. Ceci aboutit à une désorganisation globale et un dysfonctionnement des voies visuelles rétiniennes. Différentes pathologies ont été rattachées à une mutation du NR2E3 dont le syndrome d’augmentation des cônes bleus décrit par Jacobson en 1990 et le syndrome de Goldmann-Favre (forme plus sévère et plus complète avec atteinte vitréenne associée). Il s’agit des seules dystrophies rétiniennes où l’on constate une augmentation d’un type de photorécepteur.
L’atteinte associe typiquement une héméralopie suivie d’une baisse d’acuité visuelle variable avec une dégénérescence vitréorétinienne typique. La symptomatologie est bilatérale et symétrique, d’aggravation progressive. Elle peut se manifester dès l’enfance.
Segment antérieur : une cataracte sous-capsulaire postérieure, centrale, est fréquemment décrite.
Segment postérieur : l’aspect du vitré est similaire au syndrome de Wagner ou de Stickler. Il est en majeure partie liquéfié, avec la présence de condensations fibrillaires pouvant former en moyenne périphérie des rubans ou des voiles avasculaires (Fig. 15-31). Le cortex vitréen est dense et adhérent aux zones de schisis. Des amas pigmentaires nummulaires sont localisés autour du pôle postérieur, le long des arcades vasculaires. Ils ont tendance
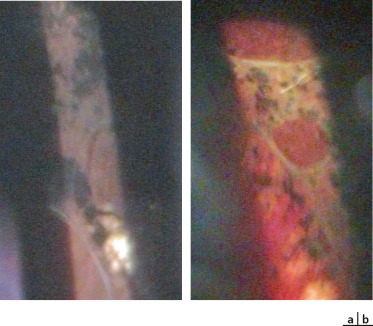
Fig. 15-31 a, b. Maladie de Goldmann-Favre avec ses amas pigmentaires et ses anomalies vitréennes.
à augmenter en taille et densité et s’étendre plus en périphérie au cours des décennies (Fig. 15-32). L’évolution se fait vers l’atrophie des photorécepteurs, d’abord périphérique (zone de concentration maximale des bâtonnets), puis centrale.
Un oedème cystoïde chronique voire un schisis fovéolaire est fréquemment retrouvé prenant l’aspect caractéristique en rayon de roue, similaire au rétinoschisis lié à l’X. Certains cas sont décrits avec un schisis plus périphérique, avec des déhiscences visibles du feuillet interne [72].
La baisse d’acuité visuelle s’explique à la fois par ces lésions « anatomiques » , mais également par le dysfonctionnement physiologique de la rétine induit par le ratio anormal des photorécepteurs (perturbation des voies ganglionnaires on/off des cônes M et L).
L’ERG est typique, avec une activité des bâtonnets très réduite voire non détectable en scotopique, une réponse atténuée identique en photopique et scotopique à un flash standard, et une réponse anormalement élevée et retardée (pic étalé) des cônes S en condition photopique lors d’une stimulation spécifique (flashes bleus). L’ERG peut devenir complètement indétectable dans les formes très avancées.
L’angiographie à la fluorescéine peut montrer une diffusion du colorant au niveau des capillaires périfovéaux, ce qui le différencie du RSJ. L’OCT est utile au diagnostic et au suivi des schisis maculaires. Dans les formes précoces, il peut montrer un épaississement de la hyaloïde postérieure et une perte du clivus fovéolaire. Le champ visuel montre des déficits variables selon la présence d’un rétinoschisis et le degré d’atrophie des photorécepteurs.
Syndrome d’augmentation des cônes bleus, dans sa forme purement rétinienne : il n’y a pas d’atteinte du vitré ni d’anomalie du segment antérieur. La sévérité du dysfonctionnement rétinien est moindre. Cependant de nombreux auteurs considèrent ces deux affections comme deux expressions phénotypiques d’un même spectre de pathologies rétiniennes liées au gène NR2E3.
Rétinoschisis lié à l’X : pathologie beaucoup plus fréquente et à transmission différente ; il n’y a pas dans ce cas d’atteinte pigmentaire périphérique, ni de diffusion à l’angiofluorographie. La baisse d’acuité est moindre, l’ERG présente un aspect « électronégatif » caractéristique.
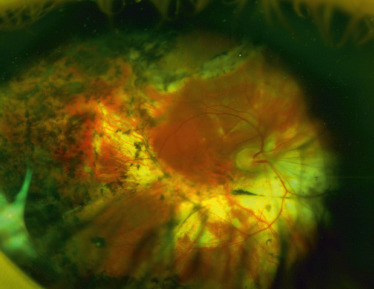
Fig. 15-32 Maladie de Goldmann-Favre stade avancé avec rétinoschisis inférieur et soulèvement rétinien nasal non évolutif (oeil adelphe perdu par DR).
Héméralopie congénitale stationnaire : la pathologie est présente dès la naissance et non évolutive. Il n’y a pas de schisis fovéolaire ou périphérique ni d’anomalie pigmentaire de la rétine. L’atteinte ERG est moins sévère avec un fonctionnement des cônes subnormal.
Rétinopathie pigmentaire : la séméiologie est très éloignée.
Il n’y a pas de traitement spécifique de la maladie. Quand un cas est suspecté, il convient de réaliser un bilan complémentaire exhaustif à la recherche des signes caractéristiques (ERG en premier lieu) avec un examen de tous les apparentés. Un bilan génétique et moléculaire spécialisé explorant le gène NR2E3 pourra être réalisé dans un centre de référence sur les rétinopathies héréditaires.
Les oedèmes maculaires chroniques peuvent être traités par inhibiteur de l’anhydrase carbonique qui aurait montré un effet bénéfique sur l’épaisseur et la sensibilité rétinienne [73]. Une chirurgie vitréorétinienne avec pelage de la membrane limitante interne pourrait diminuer les tractions s’exerçant sur la macula dans certaines formes de schisis, mais l’évolution inexorable vers l’atrophie et la dégénerescence pigmentaire limitent les indications.
P. Gastaud
Il s’agit d’une dystrophie vitréorétinienne d’évolution lente, décrite en 1974 par Hirose et al. [74], associant une liquéfaction vitréenne à des anomalies rétiniennes intéressant principalement la rétine antérieure. Ces anomalies périphériques associent des zones de « blanc sans pression » et des taches pigmentées à des punctuations jaunes et des taches blanches exsudatives donnant un aspect de flocons. La transmission de cette affection est autosomique dominante et d’expressivité variable.
La découverte de la maladie est souvent tardive, en dehors d’une recherche systématique dans le cadre d’une enquête familiale. Les premiers signes fonctionnels (baisse d’acuité visuelle, myodésopsies, signes de déchirures rétiniennes) apparaissent après l’âge de 30 ans. L’acuité visuelle reste donc maintenue, en l’absence de complications, alors que les lésions rétiniennes sont présentes très tôt dans l’enfance.
Hirose a établi une classification en quatre stades :
stade 1 : larges plages de « blanc sans pression » au niveau de la périphérie rétinienne ;
stade 2 : les ponctuations fines et claires apparaissent dans la région de l’ora serrata et des taches en flocons parsèment les zones de « blanc sans pression » . La dégénérescence fibrillaire du vitré devient visible à ce stade ;
stade 3 : apparition de mottes arrondies pigmentées, d’engainements vasculaires et d’un épaississement du vitré cortical ;
stade 4 : les pigments sont visibles sur toute la rétine antérieure et s’accompagnent de plages atrophiques chorio-épithéliales arrondies. Des condensations vitréennes et des déhiscences rétiniennes apparaissent, alors que la rétine postérieure est atteinte par des engainements vasculaires et des plages d’atrophie choroïdienne. L’apparition d’une cataracte est fréquente à ce stade vers l’âge de 40 ans.
Le bilan paraclinique a peu d’intérêt pour le diagnostic, car le bilan électrophysiologique ne s’altère que très tard dans l’évolution. Le pronostic fonctionnel est bon, en l’absence de DR secondaire : cette complication surviendrait dans un tiers des cas. Il n’existe donc aucun traitement en dehors de la prévention et du traitement chirurgical des complications rétiniennes.
Cette vitréorétinopathie très rare décrite par Hermann en 1958 [75] associe de façon inconstante une pigmentation pavimenteuse de la périphérie rétinienne à une microcornée, un glaucome et une cataracte. Il s’agit d’une affection autosomique dominante et le locus du gène a été localisé en 11q13 [76].
Cliniquement, la baisse d’acuité visuelle ne survient qu’après l’âge de 30 ans en liaison avec une cataracte précoce. Le glaucome est découvert plus tard. La maladie commence très tôt dans l’enfance, associée à une microcornée et souvent une dysmorphie faciale. L’examen du fond d’oeil met en évidence une ligne circulaire hypopigmentée séparant la rétine postérieure normale d’une rétine pré-équatoriale altérée par des pavés pigmentés, des zones atrophiques, des vaisseaux déshabités et des condensations vitréennes. L’évolution se fait par un recul de cette ligne dépigmentée, l’apparition de quelques anomalies pigmentaires proches du pôle postérieur et l’accentuation de la dystrophie vitréenne.
Au niveau des examens complémentaires, le champ visuel est initialement normal, puis s’altère avec la progression des lésions et celle des déficits glaucomateux. L’angiographie peut montrer une perméabilité anormale des vaisseaux. L’évolution se fait vers une diminution importante du champ visuel, mais la cécité est en général liée au glaucome.
Cette dystrophie vitréorétinienne, autosomique dominante et à expressivité variable, en général limitée à la périphérie rétinienne [77], a le même aspect clinique que la chorio-rétino-vitréopathie dominante autosomique. Elle ne s’en différencie que par l’absence de microcornée. L’évolution semble plus favorable et le pronostic lié surtout à l’évolution du glaucome.
Ce syndrome à transmission autosomique dominant a été décrit par Rossier et Eisner [77] à partir de l’étude d’une famille de 27 patients. L’anomalie est bilatérale, souvent associée à une myopie et caractérisée essentiellement par la présence d’une « cloison membranaire » dans la cavité vitréenne simulant un collapsus vitréen. Les auteurs signalent un taux élevé de DR secondaire.
Les dégénérescences vitréorétiniennes associées à des atteintes systémiques différentes de celle du Stickler sont [9, 78] : le syndrome de Marshall, le syndrome de Knobloch, le syndrome de Weissenbacher-Zweymuller, des vitréorétinopathies associées à des neuropathies périphériques, des malformations osseuses ou des collagénopathies de type II.
[1] Wagner H. Ein bisher unbekanntes Erbleiden des Auges (Degeneratio hyaloideoretinalis hereditaria), beobachtet im Kanton Zurich. Klin Monatsbl Augenheilkd 1938 ; 100 : 840-56.
[2] Maumenee IH, Stoll HU, Mets MB. The Wagner syndrome versus hereditary ophthalmopathy. Trans Am Ophthalmol Soc 1982 ; 80 : 349-65.
[3] Graemiger RA, Niemeyer G, Schneeberger SA, Messmer EP. Wagner vitreoretinal degeneration. Follow-up of the original pedigree. Ophthalmology 1995 ; 102 : 1830-9.
[4] Meredith SP, Richards AJ, Flanagan DW, et al. Clinical characterization and molecular analysis of Wagner syndrome. Br J Ophthalmol 2007 ; 91 : 655-9.
[5] Brown DM, Graemiger RA, Hergersberg M, et al. Genetic linkage of Wagner disease and erosive vitreoretinopathy to chromosome 5q13-14. Arch Ophthalmol 1995 ; 113 : 671-5.
[6] Miyamoto T, Inoue H, Sakamoto Y, et al. Identification of a novel splice site mutation of the CSPG2 gene in a Japanese family with Wagner syndrome. Invest Ophthalmol Vis Sci 2005 ; 46 : 2726-35.
[7] Kloeckener-Gruissem B, Amstutz C. VCAN-related vitreoretinopathy. In : GeneReviews ® [Internet]. Pagon RA, Adam MP, Ardinger HH, et al. Eds. Seattle (WA) : University of Washington, Seattle 1993–2016. 2009 Feb 3 [updated 2016 Jan 7].
[8] Rothschild PR, Brezin AP, Nedelec B, et al. A family with Wagner syndrome with uveitis and a new versican mutation. Mol Vis 2013 ; 19 : 2040-9.
[9] Gastaud P, Leguay JM, Betis F. Dégénérescences vitréorétiniennes. In : Brasseur G. Pathologie du vitré. Rapport SFO 2003. Paris : Masson ; 2003, p. 133-48.
[10] Edwards AO. Clinical features of the congenital vitreoretinopathies. Eye 2008 ; 22 : 1233-42.
[11] Stickler GB, Belau PG, Farrell FJ, et al. Hereditary progressive arthro-ophtalmolpathy. Mayo Clin Proc 1965 ; 40 : 433-55.
[12] Ahmad NN, Ala-Kokko L, Knowlton RG, et al. Stop codon in the procollagen II gene (COL2A1) in a family with the Stickler syndrome (arthro-ophthalmopathy). Proc Natl Acad Sci 1991 ; 88 : 6624-7.
[13] Richards AJ, Yates JR, Williams R, et al. A family with Stickler syndrome type 2 has a mutation in the COL11A1 gene resulting in the substitution of glycine 97 by valine in alpha 1 (XI) collagen. Hum Mol Genet 1996 ; 5 : 1339-43.
[14] Vikkula M, Mariman EC, Lui VC, et al. Autosomal dominant and recessive osteochondrodysplasias associated with the COL11A2 locus. Cell 1995 ; 80 : 431-7.
[15] Van Camp G, Snoeckx RL, Hilgert N, et al. A new autosomal recessive form of Stickler syndrome is caused by a mutation in the COL9A1 gene. Am J Hum Genet 2006 ; 79 : 449-57.
[16] Baker S, Booth C, Fillman C, et al. A loss of function mutation in the COL9A2 gene causes autosomal recessive Stickler syndrome. Am J Med Genet A 2011 ; 155 : 1668-72.
[17] Acke FR, et al. Hearing impairment in Stickler syndrome : a systematic review. Orphanet Journal of Rare Diseases 2012 ; 7 : 84.
[18] Richards AJ, Martin S, Yates JR, et al. COL2A1 exon 2 mutations : relevance to the Stickler and Wagner syndromes. Br J Ophthalmol 2000 ; 84 : 364-71.
[19] Snead MP, Payne SJ, Barton DE, et al. Stickler syndrome : correlation between vitreoretinal phenotypes and linkage to COL2A1. Eye 1994 ; 8 : 609-14.
[20] Snead MP, Yates JRW, Pope FM, et al. Masked confirmation of linkage between type 1 congenital vitreous anomaly and col 2A1 in Stickler syndrome. Graefe’s Arch Clin Exp Ophthalmol 1996 ; 234 : 720-1.
[21] Vintiner GM, Temple IK, Middleton-Price HR, et al. Genetic and clinical heterogeneity of Stickler syndrome. Am J Med Genet 1991 ; 41 : 44-8.
[22] Ahmad NN, McDonald-McGinn DM, Dixon P, et al. PCR assay confirms diagnosis in syndrome with variably expressed phenotype : mutation detection in Stickler syndrome. J Med Genet 1996 ; 33 : 678-81.
[23] Perveen R, Hart-Holden N, Dixon MJ, et al. Refined genetic and physical localisation of the Wagner disease (WGN1) locus and the genes CRTL1 and CSPG2 to a 2- to 2.5-cM region of chromosome 5q14.3. Genomics 1999 ; 57 : 219-26.
[24] Brunner HG, Van Beersum SEC, Warman ML, et al. A Stickler syndrome gene is linked to chromosome 6 near the COL11A2 gene. Hum Mol Genet 1994 ; 3 : 1561-4.
[25] Richards AJ, Baguley DM, Yates JR, et al. Variation in the vitreous phenotype of Stickler syndrome can be caused by different amino acid substitutions in the X position of the type II collagen Gly-X-Y triple helix. Am J Hum Genet 2000 ; 67 : 1083-94.
[26] Betis F, Hofman P, Gastaud P. Vitreous changes in Stickler syndrome. J Fr Ophtalmol 2003 ; 26 : 386-90.
[27] Yokoi T, Koide R, Matsuoka K, et al. Analysis of the vitreous membrane in a case of type 1 Stickler syndrome. Graefes Arch Clin Exp Ophthalmol 2009 ; 247 : 715-8.
[28] MacRae ME, Patel DV, Richards AJ, et al. Type 1 Stickler syndrome : a histological and ultrastructural study of an untreated globe. Eye 2006 ; 20 : 1061-7.
[29] Miyashita K, Tokunaga M, Akiyama K, et al. Electron microscopic study on the vitreous membrane of the Stickler syndrome. Nippon Ganka Gakkai Zasshi 1994 ; 98 : 86-91.
[30] Seery CM, Pruett RC, Liberfarb RM, et al. Distinctive cataract in the Stickler syndrome. Am J Ophthalmol 1990 ; 110 : 143-8.
[31] Richards AJ, McNinch A, Martin H, et al. Stickler syndrome and the vitreous phenotype : mutations in COL2A1 and COL11A1. Hum Mutat 2010 ; 31 : E1461–E1471.
[32] Richards AJ, Laidlaw M, Whittaker J, et al. High efficiency of mutation detection in type 1 stickler syndrome using a two-stage approach : vitreoretinal assessment coupled with exon sequencing for screening COL2A1. Hum Mutat 2006 ; 27 : 696-704.
[33] Parma ES, Körkkö J, Hagler WS, et al. Radial perivascular retinal degeneration : a key to the clinical diagnosis of an ocular variant of Stickler syndrome with minimal or no systemic manifestations. Am J Ophthalmol 2002 ; 134 : 728-34.
[34] Vu CD, Brown J Jr, Körkkö J, et al. Posterior chorioretinal atrophy and vitreous phenotype in a family with Stickler syndrome from a mutation in the COL2A1 gene. Ophthalmology 2003 ; 110 : 70-7.
[35] Snead MP, McNinch AM, Poulson AV, et al. Stickler syndrome, ocular-only variants and a key diagnostic role for the ophthalmologist. Eye 2011 ; 25 : 1389-400.
[36] Szymko-Bennett YM, Mastroianni MA, Shotland LI, et al. Auditory dysfunction in Stickler syndrome. Arch Otolaryngol Head Neck Surg 2001 ; 127 : 1061-8.
[37] Monin CL, Van Effenterre G, Andre-Sereys P, et al. Prophylaxie du décollement de rétine de la maladie de Wagner-Stickler. Etude comparative des différentes méthodes. A propos de vingt deux cas. J Fr Ophtalmmol 1994 ; 17 : 167-74.
[38] Carroll C, Papaioannou D, Rees A, et al. The clinical effectiveness and safety of prophylactic retinal interventions to reduce the risk of retinal detachment and subsequent vision loss in adults and children with Stickler syndrome : a systematic review. Health Technol Assess 2011 ; 15 : 1-62.
[39] Fincham GS, Pasea L, Carroll C, et al. Prevention of retinal detachment in Stickler syndrome : the Cambridge prophylactic cryotherapy protocol. Ophthalmology 2014 ; 121 : 1588-97.
[40] Abeysiri P, Bunce C, da Cruz L. Outcomes of surgery for retinal detachment in patients with Stickler syndrome : a comparison of two sequential 20-year cohorts. Graefes Arch Clin Exp Ophthalmol 2007 ; 245 : 1633-8.
[41] Errera MH, Liyanage SE, Moya R, et al. Primary scleral buckling for pediatric rhegmatogenous retinal detachment. Retina 2015 ; 35 : 1441-9.
[42] Haas J. Über das Zusammenvorkommen von Veranderugen der Retina und Choroidea. Arch Augenheikd 1898 ; 37 : 343-8.
[43] Tantri A, Vrabec TR, Cu-Unjieng A, et al. X-Linked retinoschisis : a clinical and molecular genetic review. Surv Ophthalmol 2004 ; 49 : 214-30.
[44] TaninoT, Katsumi O, Hirose T. Electrophysiological similarities between two eyes with X-linked recessive retinoschisis. Doc Ophthalmol 1985 ; l60 : 149-61.
[45] George NDL, Yates JRW, Moore AT. Clinical features in affected males with X-linked retinoschisis. Arch Ophthalmol 1996 ; 114 : 274-80.
[46] Sieving PA, MacDonald IM, Chan S. X-Linked Juvenile Retinoschisis. In : GeneReviews ® [Internet]. Pagon RA, Adam MP, Ardinger HH, et al. Eds. Seattle (WA) : University of Washington, Seattle 1993–2016. 2003 Oct 24 [updated 2014 Aug 28].
[47] Apushkin MA, Fishman GA, Rajagopalan AS. Fundus findings and longitudinal study of visual acuity loss in patients with X-linked retinoschisis. Retina 2005 ; 25 : 612-8.
[48] Yang HS, Lee JB, Yoon YH, Lee JY. Correlation between spectral-domain OCT findings and visual acuity in X-linked retinoschisis. Invest Ophthalmol Vis Sci 2014 ; 55 : 3029-36.
[49] Al-Swaina N, Nowilaty SR. Macular hole in juvenile X-linked retinoschisis. Saudi J Ophthalmol 2013 ; 27 : 283-6.
[50] Moldaya RS, Kellnerb U, Weberc BH. X-linked juvenile retinoschisis : clinical diagnosis, genetic analysis, and molecular mechanisms. Prog Retin Eye Res 2012 ; 31 : 195-212.
[51] Vincent A, Robson AG, Neveu MM, et al. A phenotype–genotype correlation study of X–Linked retinoschisis. Ophthalmology 2013 ; 120 : 1454-64.
[52] Bush RA, Wei LL, Sieving PA. Convergence of human genetics and animal studies : gene therapy for X-linked retinoschisis. Cold Spring Harb Perspect Med 2015 ; 5 : a017368.
[53] Study of RS1 ocular gene transfer for X-linked retinoschisis ; ClinicalTrials.gov Identifier : NCT02317887.
[54] Safety and Efficacy of rAAV-hRS1 in Patients with X-linked Retinoschisis (XLRS) ; ClinicalTrials.gov Identifier : NCT02416622.
[55] Yu H, Li T, Luo Y, et al. Long-term outcomes of vitrectomy for progressive X-linked retinoschisis. Am J Ophthalmol 2012 ; 154 : 394-402.
[56] Ferrone PJ, Trese MT, Lewis H. Vitreoretinal surgery for complications of congenital retinoschisis. Am J Ophthalmol 1997 ; 123 : 742-7.
[57] Rosenfeld PJ, Flynn HW Jr, McDonald HR, et al. Outcomes of vitreoretinal surgery in patients with X-linked retinoschisis. Ophthalmic Surg Lasers 1998 ; 29 : 190-7.
[58] Trese MT, Ferrone PJ. The role of inner wall retinectomy in the management of juvenile retinoschisis. Graefe’s Arch Clin Exp Ophthalmol 1995 ; 233 : 706-8.
[59] George NDL, Yates JRW, Moore AT. Clinical features in affected males with X-linked retinoschisis. Arch Ophthalmol 1996 ; 114 : 274-80.
[60] Sikkink SK, Biswas S, Parry NR, et al. X-Linked retinoschisis : an update. J Med Genet 2007 ; 44 : 225-32.
[61] Pendergast SD, Trese MT. Familial exudative vitreoretinopathy : results of surgical management. Ophthalmology 1998 ; 105 : 1015-23.
[62] Gilmour DF. Familial exudative vitreoretinopathy and related retinopathies. Eye 2015 ; 29 : 114.
[63] Finis D, Stammen J, Joussen AM. Familial exudative vitreoretinopathy. Ophthalmologe 2010 ; 107 : 683-91.
[64] Chen SN, Hwang JF, Lin CJ. Clinical characteristics and surgical management of familial exudative vitreoretinopathy-associated rhegmatogenous retinal detachment. Retina 2012 ; 32 : 220-5.
[65] Van Nouhuys CE. Signs, complications, and platelet aggregation in familial exudative vitreoretinopathy. Am J Ophthalmol 1991 ; 111 : 3441.
[66] Brown DM, Kimura AE, Weingeist TA, Stone EM. Erosive vitreoretinopathy. A new clinical entity. Ophthalmology 1994 ; 101 : 694-704.
[67] Mukhopadhyay A, Nikopoulos K, Maugeri A, et al. Erosive vitreoretinopathy and wagner disease are caused by intronic mutations in CSPG2/Versican that result in an imbalance of splice variants. Invest Ophthalmol Vis Sci 2006 ; 47 : 3565-72.
[68] Brown DM, Graemiger RA, Hergersberg M, et al. Genetic linkage of Wagner disease and erosive vitreoretinopathy to chromosome 5q13-14. Arch Ophthalmol 1995 ; 113 : 671-5.
[69] Favre M. Two cases of hyaloid‑retinal degeneration. Ophthalmologica 1958 ; 135 : 604-9.
[70] Hamel C. Goldmann-Favre syndrome. ORPHA53540. Orphanet 2009.
[71] Milam AH, Rose L, Cideciyan AV, et al. The nuclear receptor NR2E3 plays a role in human retinal photoreceptor differentiation and degeneration. Proc Natl Acad Sci U S A 2002 ; 99 : 473-8.
[72] Tang S, Ding X, Luo Y. Hereditary vitreoretinal degenerations. In : Ryan SJ. Retina. Vol. 2. 5th ed. Elsevier Saunders ; 2013, p. 836-48.
[73] Genead MA, Fishman GA, McAnany JJ. Efficacy of topical dorzolamide for treatment of cystic macular lesions in a patient with enhanced S-cone syndrome. Doc Ophthalmol 2010 ; 121 : 231-40.
[74] Hirose T, Lee KY, Schepens CL. Snowflake degeneration in hereditary vitreoretinal degeneration. Am J Ophthalmol 1974 ; 77 : 143-53.
[75] Hermann P. Le syndrome microphtalmie-rétinite pigmentaire-glaucome. Arch Ophtalmol 1958 ; 18 : 17-24.
[76] Yardley J, Leroy BP, Hart-Holden N, et al. Mutations of VMD2 splicing regulators cause nanophthalmos and autosomal dominant vitreoretinochoroidopathy (ADVIRC). Invest Ophthalmol Vis Sci 2004 ; 45 : 3683-9.
[77] Rossier J, Eisner G. The pseudo-posterior limiting layer syndrome : a vitreoretinal heredodegeneration with autosomal dominant transmission. Graefe’s Arch Clin Exp Ophthalmol 1994 ; 232 : 16-24.
[78] Dufier JL, Kaplan J : OEil et génétique. Paris : Masson ; 2005, p. 339-59.
F. Matonti , J. Benichou
L’hémorragie intravitréenne de l’enfant est une pathologie rare. Si, chez l’adulte, les étiologies, les manifestations cliniques et la prise en charge sont bien documentées, les données chez l’enfant sont moins nombreuses et la conduite à tenir moins bien codifiée.
Chez le grand enfant, la baisse d’acuité visuelle constitue le signe d’appel le plus fréquent. Tout l’enjeu diagnostique se trouve à l’âge préverbal : un strabisme, un nystagmus ou un simple changement de comportement (désintérêt pour l’environnement, nonreconnaissance des visages familiers) peuvent être les seuls signes d’appel [1, 2]. L’examen clinique doit s’attacher à rechercher l’uniou la bilatéralité du saignement, les antécédents ainsi que la présence d’éventuels signes systémiques associés.
Avant l’âge de 1 an, les hémorragies intravitréennes bilatérales sont le plus souvent secondaires à un syndrome du bébé secoué [1]. Cette affection grave est occasionnée par des mouvements d’accélération/décélération lors de secousses manuelles violentes qui entraînent des lésions neurologiques ainsi que vasculaires. Elle est évoquée devant la présence d’hématomes cérébraux, en général sous-duraux, d’hémorragies intra-oculaires (rétiniennes et vitréennes) et l’absence de signes extérieurs de traumatisme. Typiquement, les hémorragies rétiniennes sont bilatérales, disséminées et peuvent être pré-, intra- ou plus rarement sous-rétiniennes. Le pronostic visuel serait plus sombre et les atteintes neurologiques plus importantes en cas d’hémorragie intravitréenne [3].
En dehors de ce contexte, les causes traumatiques restent les plus fréquentes (70 % des cas environ) : traumatisme accidentel perforant ou non perforant, traumatisme néonatal, syndrome de Silverman [1, 4].
Parmi les causes non traumatiques, on distingue principalement les hémorragies intravitréennes survenant dans le cadre d’une
Fig. 15-33 Arbre diagnostique présentant les principales étiologies d’hémorragie intravitréenne chez l’enfant.
ROP : rétinopathie du prématuré ; VREF : vitréorétinopathie exsudative familiale.
rétinopathie des prématurés, celles secondaires aux vascularites, aux maladies hématologiques (leucémie, drépanocytose, etc.), aux vitréorétinopathies exsudatives familiales, aux rétinopathies diabétiques, aux tumeurs oculaires, aux déchirures rétiniennes. De nombreuses hémorragies restent cependant idiopathiques [2, 5].
Diverses complications [4, 6] peuvent grever le pronostic d’une hémorragie intravitréenne de l’enfant. On peut distinguer les complications fonctionnelles (amblyopie profonde en cas d’hémorragie dense, strabisme, myopie par privation visuelle) et les complications anatomiques (DR, membrane épirétinienne).
Le vitré de l’enfant est beaucoup plus cohérent et gélatineux que chez l’adulte : la résorption spontanée d’une hémorragie intravitréenne est donc plus lente. La vitrectomie doit être précoce [7], dès la troisième semaine qui suit le saignement afin d’éviter les différentes complications possibles.
Les principales étiologies d’hémorragie intravitréenne chez l’enfant sont présentées dans la figure 15-33.
[1] Spirn MJ, Lynn MJ, Hubbard GB 3rd. Vitreous hemorrhage in children. Ophthalmology 2006 ; 113 : 848-52.
[2] Sudhalkar A, Chhablani J, Jalali S, et al. Spontaneous vitreous hemorrhage in children. Am J Ophthalmol 2013 ; 156 : 1267-71.e2.
[3] Matthews GP, Das A. Dense vitreous hemorrhages predict poor visual and neurological prognosis in infants with shaken baby syndrome. J Pediatr Ophthalmol Strabismus 1996 ; 33 : 260‑5.
[4] Gastaud P, Chaudoutaud E. Hémorragies intra-vitréennes. In : Brasseur G. Pathologie du vitré. Rapport SFO 2003. Paris : Masson ; 2003, p. 277-95.
[5] Sudhalkar A, Chhablani J, Rani PK, et al. Bilateral vitreous hemorrhage in children : clinical features and outcomes. J Ophthalmic Vis Res 2015 ; 10 : 139-43.
[6] Miller-Meeks MJ, Bennett SR, Keech RV, Blodi CF. Myopia induced by vitreous hemorrhage. Am J Ophthalmol 1990 ; 109 : 199‑203.
[7] Ferrone PJ, de Juan E Jr. Vitreous hemorrhage in infants. Arch Ophthalmol 1994 ; 112 : 1185-9.
Pour tout le chapitre, les figures proviennent de la collection de P. Gastaud ou de F. Metge-Galatoire, sauf mention contraire.