Pathologie du nerf optique
Coordonné par P. Lebranchu
M. Robert
Par pathologie développementale on entend « pathologie malformative » (donc par définition congénitale) au sens large : non seulement dysgénésique (résultant d'une anomalie génétique ou cytogénétique), mais aussi disruptive ou clastique (résultant d'un « accident » au cours de la grossesse) ou encore déformative (résultant d'une déformation, au sens de David Smith, au cours de la grossesse).
Il s'agit de l'anomalie développementale du nerf optique la plus fréquente. Le mode de révélation d'une hypoplasie papillaire est variable. Quand elle est bilatérale, le tableau est celui d'un retard de développement de la fonction visuelle chez le nourrisson, avec nystagmus et malvoyance de degré variable. Quand elle est unilatérale, l'amblyopie organique est le plus souvent révélée par un strabisme; le diagnostic n'est cependant fait qu'à l'âge de la chirurgie du strabisme.
L'aspect clinique est typique : la papille est de petite taille, de coloration variable (blanche, jaune ou rose), souvent entourée d'un liseré blanc jaunâtre (correspondant au canal scléral vide), lui-même bordé d'un trait hypo- ou hyperpigmenté (aspect dit de papille en cocarde) (fig. 21-1a). Les veines rétiniennes de premier ordre sont généralement très tortueuses, ce qui est une aide au diagnostic lors de l'examen du fond d'œil d'un nourrisson nystagmique (fig. 21-1a et b); plus rarement les vaisseaux rétiniens de premier ordre peuvent au contraire apparaître trop rectilignes (fig. 21-1c); souvent enfin, le branchement des vaisseaux rétiniens est inférieur à la normale (fig. 21-1a et c), sans que l'on sache si ces anomalies de branchement sont corrélées aux insuffisances hormonales [1].
La définition de l'hypoplasie papillaire est matière à débat : certains auteurs ont proposé que celle-ci repose sur le rapport entre la taille de la papille et la distance fovéa-papille, d'autres ont proposé de considérer le diamètre de la papille [2, 3]. Suivre cette définition en clinique revient à qualifier des petites papilles à fonction visuelle normale d'hypoplasiques et donc logiquement à réaliser dans ces cas-là le même bilan (alors invariablement normal) que dans les cas correspondant aux formes cliniques typiques. Nous sommes en faveur d'une acception restrictive du terme « hypoplasie papillaire » , réservé à des papilles non seulement morphologiquement, mais aussi fonctionnellement anormales [4]. Nous qualifions les autres de « petites papilles constitutionnelles » , sans parler d'hypoplasie.
La physiopathologie des hypoplasies papillaires n'est pas entièrement élucidée : dans certains cas, il s'agirait d'une faillite du développement axonal; dans d'autres, d'une apoptose excessive des axones du nerf optique au cours du deuxième trimestre de grossesse. Les principaux facteurs de risque seraient la primiparité et le jeune âge de la mère [5]. Des associations à des syndromes cytogénétiques et à des expositions à des toxiques pendant la grossesse ont été rapportées.
La présence d'une hypoplasie papillaire typique au fond d'œil signe celle d'une hypoplasie du nerf optique depuis la lame criblée jusqu'au chiasma (fig. 21-2), celui-ci étant généralement également hypoplasique (hémi-hypoplasique ou entièrement hypoplasique selon le caractère uni- ou bilatéral de l'hypoplasie papillaire). Le nerf optique apparaît donc de diamètre plus petit que la normale, ce qui est souvent qualifié à tort d' « atrophie » du nerf optique. Il n'existe en effet aucun moyen de différencier sur une imagerie par résonance magnétique (IRM) unique une hypoplasie d'une atrophie du nerf optique (seule l'évolutivité documentée, sur deux IRM successives, d'une diminution de diamètre du nerf optique permet de parler d'atrophie); c'est l'examen clinique de la papille optique qui permet de faire cette distinction.
Les hypoplasies papillaires, qu'elles soient uni- ou bilatérales, peuvent être soit isolées, soit associées à d'autres malformations du système nerveux central. La désormais classique « dysplasie septo-optique » de de Morsier (hypoplasie papillaire, absence de septum pellucidum et agénésie du corps calleux) n'est qu'une des présentations possibles des hypoplasies papillaires syndromiques : des ectopies hypophysaires et des anomalies hémisphériques (anomalies de migration ou anomalies disruptives) sont présentes dans la moitié des cas environ (fig. 21-3). La présence d'un hypopituitarisme associé est fréquente (15 à 70 % selon les études); elle n'est cependant pas toujours associée à une ectopie hypophysaire visible sur l'IRM et doit donc être recherchée indépendamment et systématiquement [6]. Les troubles du rythme veille-sommeil et les troubles du développement (plus fréquents en cas d'anomalies du corps calleux) sont aussi fréquents et doivent être recherchés et pris en charge précocement [7, 8]. De façon exceptionnelle, les hypoplasies papillaires peuvent s'associer à des tumeurs congénitales des voies optiques, pouvant entraîner une atrophie optique surajoutée sur une papille hypoplasique ou sur une papille saine controlatérale dans le cas d'une forme unilatérale [9].
Le bilan de première intention à réaliser devant une hypoplasie papillaire est résumé dans le Tableau 21-1.
Il n'existe à l'heure actuelle pas de traitement des hypoplasies papillaires qui permettrait un gain de fonction visuelle. La part fonctionnelle de l'amblyopie organique résultant d'une hypoplasie papillaire unilatérale doit être prise en compte et rééduquée de façon pragmatique. Les tentatives de thérapie par des cellules souches se sont jusqu'à présent soldées par des échecs [10], comportent des risques non maîtrisés et leur rapport bénéfice/risque plaide très largement, en 2017, contre leur réalisation.
Elles ne sont pas rares. Le diagnostic est posé soit lors de l'examen systématique d'un enfant né prématurément, soit lors d'un bilan de malvoyance. Ici encore, nous sommes en faveur de l'emploi du terme « hypoplasie » dans son sens le plus restrictif :
- – en cas de fonction visuelle normale, les papilles excavées sont dites « papilles excavées constitutionnelles » : elles sont découvertes lors d'un examen systématique chez un enfant et le principal diagnostic différentiel est le glaucome juvénile débutant;
- – en cas de malvoyance, l'hypoplasie papillaire peut ou non s'accompagner de nystagmus en fonction précisément de l'importance de l'excavation (la malvoyance résulte principalement des lésions associées des radiations optiques associées à la leucomalacie périventriculaire, tandis que la présence d'un nystagmus dépend de l'importance de l'hypoplasie papillaire; l'importance de l'hypoplasie n'est pas strictement corrélée aux lésions des radiations, elle dépend aussi du moment de la grossesse ou les phénomènes d'hypoxo-ischémies sont survenus).
L'aspect clinique est très différent de celui des autres hypoplasies papillaires : il s'agit d'une papille de taille normale, avec une excavation centrée, bilatérale et assez symétrique (fig. 21-4). Elle pourrait être prise pour une papille glaucomateuse, mais d'une part il n'existe aucun autre signe de glaucome, d'autre part le contexte (prématurité) est évocateur [11]. Quand l'enfant grandit, il est parfois possible de réaliser un champ visuel, qui montre le plus souvent un déficit inférieur bilatéral [12].
Cette anomalie est classiquement associée à la leucomalacie périventriculaire de la prématurité, qui elle-même s'inscrit le plus souvent dans un tableau de malvoyance congénitale dite sous-corticale. On peut cependant aussi l'observer chez des enfants nés prématurément, ayant présenté un retard de maturation visuelle, avec un degré variable de leucomalacie périventriculaire, mais dont la fonction visuelle est bonne.
Le mécanisme résulterait de la dégénérescence transsynaptique rétrograde depuis les axones des radiations optiques – lésés par hypoxie-ischémie entre la fin du deuxième trimestre et le début du troisième trimestre – jusqu'à la papille optique dont le diamètre à ce stade serait déjà normal.
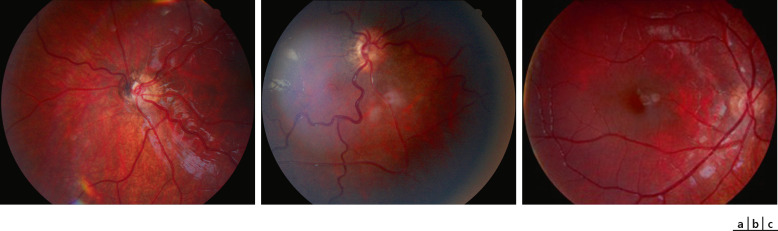
Fig. 21-1 Hypoplasie papillaire typique.
a. Hyperpigmentations péripapillaires ; tortuosité des veines de premier ordre. Branchements vasculaires inférieurs à la normale. b. Hypoplasie papillaire typique ; hyper- et hypopigmentations péripapillaires ; tortuosité des veines de premier ordre. c. Hypoplasie papillaire typique ; hypopigmentation péripapillaire ; rectitude des veines de premier ordre ; branchements vasculaires inférieurs à la normale.

Tableau 21-1 Bilan recommandé devant une hypoplasie papillaire.
FSH : folliculo-stimulating hormone ; IGF1 : insulin-like growth factor 1 ; IRM : imagerie par résonance magnétique ; LH : luteinising hormone ; T4us : hormone thyroïdienne de type 4 ; TSH : thyroid stimulating hormone ;
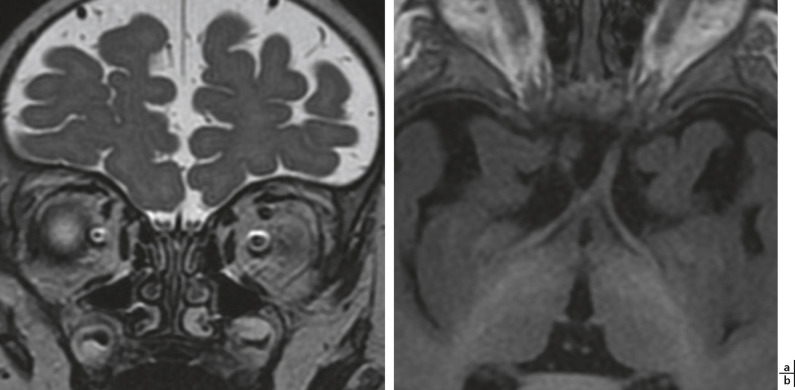
Fig. 21-2 Hypoplasie du nerf optique droit.
a. Séquence T2, plan coronal : l’hypoplasie est nette au niveau de l’orbite. b. Séquence T1, plan neuro-oculaire : l’hypoplasie se prolonge jusqu’au chiasma optique.
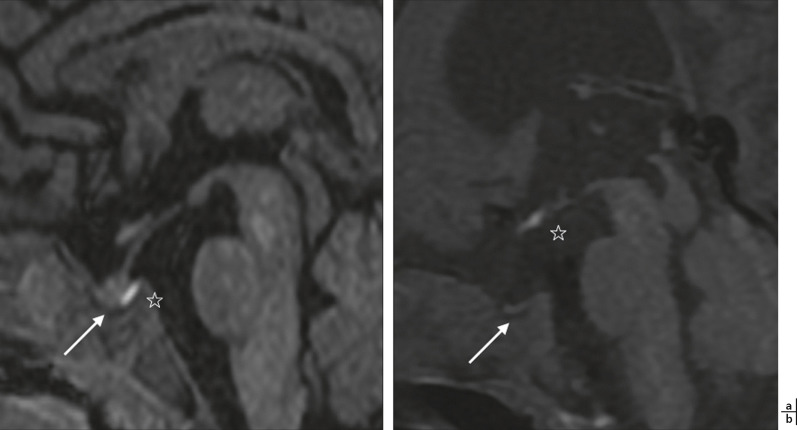
Fig. 21-3 Ectopie pituitaire.
a. Post-hypophyse (hyperintense, à côté de l’étoile) normale, en place, dans la selle turcique (flèche), sous une tige infundibulaire normale. b. Post-hypophyse (hyperintense, à côté de l’étoile) ectopique, très au-dessus de la selle turcique (flèche), sans tige infundibulaire visualisable.
Le contexte de prématurité étant connu avant le diagnostic, il est rare qu'une imagerie cérébrale n'ait pas été réalisée dans les cas de malvoyance associée; dans les autres cas, elle peut montrer des anomalies variables de la substance blanche. Aucun bilan biologique spécifique n'est recommandé.
Trois types principaux existent : hypoplasie supérieure, hypoplasie hémioptique de type chiasmatique et hypoplasie hémioptique homonyme de type rétrochiasmatique. Il s'agit souvent d'une découverte fortuite. Les colobomes papillaires (qui représentent certes la principale cause d'hypoplasie papillaire) sont traités à part.
Les hypoplasies papillaires supérieures (topless Disk) sont classiquement observées chez les enfants nés de mères diabétiques insulino-dépendantes (incidence environ 8 % ) [13]. L'émergence des vaisseaux se fait à la partie supérieure de la papille optique, dont la portion inférieure apparaît normale (fig. 21-5a). Il peut exister un croissant de canal scléral « vide » au-dessus de la papille optique. Elles s'accompagnent d'un déficit campimétrique altitudinal inférieur. Elles sont paradoxalement généralement isolées. Elles peuvent aussi être observées en dehors du contexte de diabète maternel [14] et ne requièrent a priori pas de bilan spécifique.
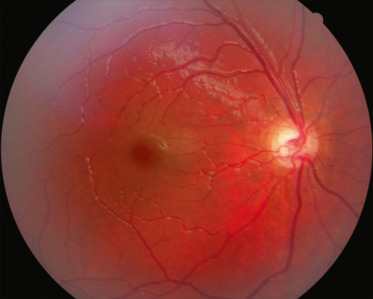
Fig. 21-4 Hypoplasie papillaire excavée liée à la prématurité, associée à une leucomalacie périventriculaire.
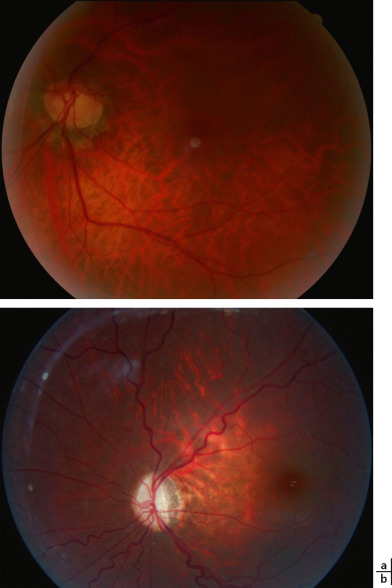
Fig. 21-5 Hypoplasie papillaire segmentaire.
a. Supérieure (topless disc) chez un sujet né de mère diabétique insulino-dépendante.b. Hémioptique en bande.
Les hypoplasies papillaires hémioptiques de type chiasmatique résultent d'une malformation constitutionnelle du chiasma (achiasmie, avec alors nystagmus de type seesaw) ou d'une lésion compressive intra-utérine (gliome, ou plus rarement kyste de la poche de Rathke, avec parfois peu ou pas de symptômes) [15]. Elles se caractérisent par une hypoplasie en bande (ou bow tie) bilatérale (fig. 21-5b), par atteinte des fibres ganglionnaires croisées, similaire à l'atrophie papillaire en bande bilatérale des lésions chiasmatiques acquises. Elles imposent la réalisation d'une imagerie cérébrale.
Les hypoplasies papillaires hémioptiques homonymes de type rétrochiasmatique résultent d'une lésion congénitale, le plus souvent disruptive, des voies visuelles rétrogéniculées. Elles se caractérisent par une hypoplasie en bande (évidente cliniquement) du côté de la lésion par atteinte des fibres ganglionnaires croisées et par une hypoplasie en sablier par atteinte des fibres ganglionnaires directes (le plus souvent difficile à mettre en évidence cliniquement, mais bien visible en tomographie par cohérence optique) controlatérale à la lésion, tout comme les atrophies papillaires sectorielles des lésions acquises des bandelettes optiques (fig. 21-6) [16].
Les colobomes, du grec koloboma (mutilé), désignent stricto sensu un groupe de malformations correspondant à une « fissure » de l'œil. Ils résultent d'un défaut de fermeture de la fente embryonnaire; ce défaut survient entre le début de l'invagination linéaire ventrale simultanée de la vésicule optique et du pédicule optique à la 5e semaine de vie intra-utérine et la fermeture de la fente embryonnaire qui résulte de cette invagination à la 7e semaine de vie intra-utérine. Lorsque le défaut concerne la portion la plus postérieure de la fente, le défect est papillaire. Par souci de synthèse, les colobomes choriorétiniens sont traités ici également.
Les conditions du diagnostic sont éminemment variables : depuis un syndrome du nystagmus précoce avec comportement de malvoyance pour les très vastes colobomes choriorétiniens ou papillaires, à une découverte fortuite pour les plus petits, en passant par un strabisme précoce secondaire à une amblyopie organique unilatérale pour les formes asymétriques. Le diagnostic est clinique et le plus souvent évident : l'aspect du colobome est celui de la sclère, sur laquelle cheminent les vaisseaux « rétiniens » ; à la jonction avec la rétine normale se trouve un liseré pigmenté souvent irrégulier et de largeur variable. Les zones colobomateuses sont déformées en un staphylome postérieur localisé bien visible en imagerie (échographie B, scanner ou IRM orbitaire). La clé du diagnostic réside dans la localisation : inféromédiane (sur le méridien de 5 heures à l'œil droit et celui de 7 heures à l'œil gauche, voir (fig. 21-7). Les lésions ayant l'aspect d'un colobome mais qui ne sont pas situées sur ce méridien de fermeture de la fente embryonnaire ne sont donc pas des colobomes, contrairement à une certaine tradition remontant à von Szily et qui tendrait à qualifier toutes les malformations du nerf optique de colobome. Les diagnostics différentiels comportent : les cicatrices de toxoplasmose (et d'autres agents infectieux) pour les colobomes choriorétiniens; les excavations papillaires constitutionnelles, les papilles en fleur de liseron et les papilles du syndrome rein-colobome (dans les deux cas l'excavation est centrée) pour les colobomes papillaires. Il est important de documenter le colobome par des photographies et de le classer (fig. 21-7 et fig. 21-8). La classification la plus communément employée reste celle d'Ida Mann (fig. 21-9) [17]. Nous lui adjoignons systématiquement celle de Gopal (Tableau 21-2) [18], dont l'intérêt est pronostique. Il semble que ni l'étendue ni la localisation du colobome présagent en revanche de son caractère syndromique.
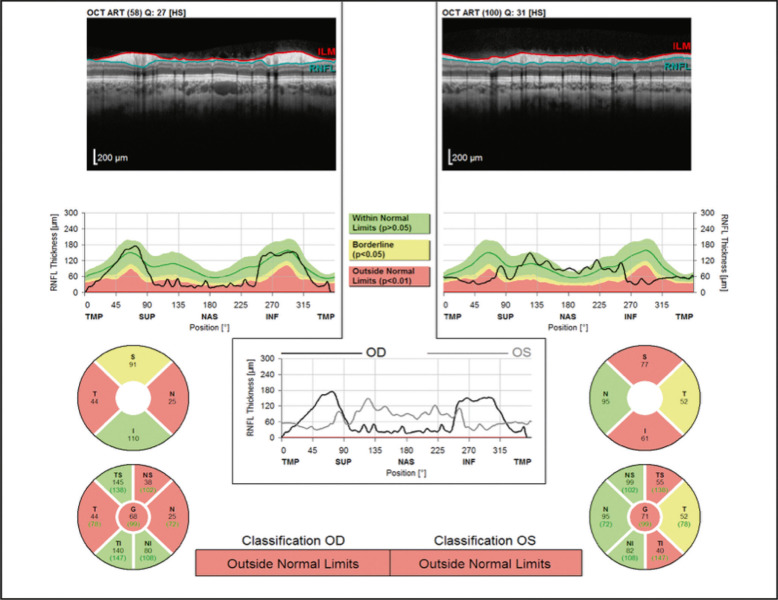
Fig. 21-6 Aspect en OCT d'une hypoplasie papillaire en bande à droite et en sablier à gauche, réalisant une hypoplasie papillaire hémioptique homonyme de type rétrochiasmatique, ici gauche (lésion congénitale de la bandelette optique gauche).
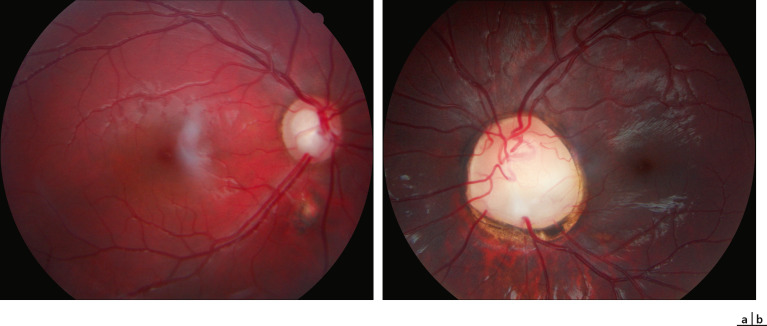
Fig. 21-7 Colobome papillaire type 4 d’Ida Mann.
a. A minima : papille de grande taille et excavation excentrée à 5 heures associées à un colobome choriorétinien du type 7 d’Ida Mann (dit rudimentaire), sous la forme d’une simple anomalie de pigmentation dans l’aire de fermeture de la fente embryonnaire. b. De taille intermédiaire.
Plusieurs syndromes comportant un colobome sont décrits. Au premier rang sans doute, le syndrome CHARGE (Coloboma, Heart defect, Atresia choanae, Retarded growth, Genital anomalies, Ear anomalies) [19]. Plusieurs définitions cliniques faisant appel à des critères existent [20, 20]; la plus utilisée actuellement date de 2005 (Tableau 21-3); le colobome compte au rang des critères majeurs et est présent dans la majorité des cas; le diagnostic est généralement suspecté soit en période prénatale, soit en service de néonatologie ou de pédiatrie. Le syndrome résulte d'un dysfonctionnement au cours du deuxième mois de gestation. Une mutation du gène CHD7 codant un acide désoxyribonucléique (ADN) hélicase à chromodomaine est retrouvée dans plus de 75 % des cas. Le colobome peut intéresser l'iris, la choriorétine ou la papille.
Le syndrome du cat eye tire son nom de l'aspect que l'œil confère à des patients atteints un colobome irien; celui-ci constitue l'une des trois malformations de la triade qui définit classiquement le syndrome, avec une atrésie anale et des anomalies pré-auriculaires (sinus ou tubercules prétragiens). Peuvent s'y associer une cardiopathie, des anomalies du tractus urinaire ou du squelette, un déficit intellectuel généralement modéré. L'hétérogénéité clinique est importante et seuls 41 % des patients présentent la triade phénotypique classique. Dans environ 80 % des cas, le caryotype montre la présence d'un petit chromosome surnuméraire dérivé du chromosome 22 (trisomie ou tétrasomie 22 proximale) [22, 23].

Tableau 21-2 Classification de Gopal des colobomes choriorétiniens (1996).
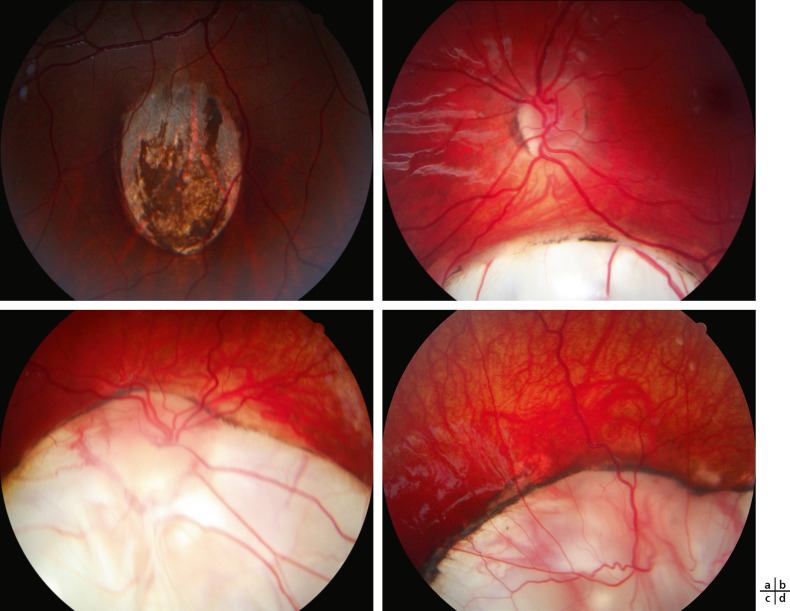
Fig. 21-8 Colobome choriorétinien.
a. Type 5 d’Ida Mann. b. Type 3 d’Ida Mann et colobome papillaire (type 4 d’Ida Mann) : type III de Gopal. c. Type 2 d’Ida Mann et type V de Gopal. d. Type 1 d’Ida Mann et type VI de Gopal.
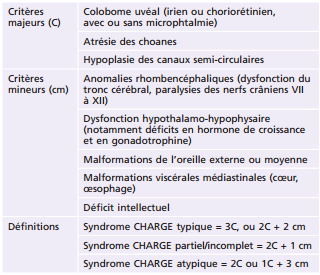
Tableau 21-3 Critères diagnostiques du syndrome CHARGE d'après Verloes [21].
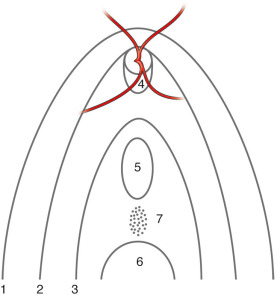
Fig. 21-9 Classification des colobomes papillaires et choriorétiniens selon Ida Mann (1937).
Chaque numéro correspond à un type. 1. Le colobome englobe la papille et s’étend au-dessus d’elle. 2. Le colobome englobe la papille mais ne la dépasse pas. 3. Ces deux variétés peuvent présenter un pont de tissu normal qui divise le colobome en deux parties, une partie supérieure et une partie inférieure ; c’est le colobome à pont. 4. Le colobome est localisé au bord inférieur de la papille sous forme d’un croissant. 5. Le colobome n’atteint ni la papille ni la périphérie et se présente sous forme d’une tache blanchâtre ovalaire. 6. Le colobome est tout à fait périphérique. 7. Le colobome est rudimentaire, il n’existe qu’une traînée pigmentaire située dans la direction de la fente foetale.
Les colobomes uvéaux ont été rapportés dans : de nombreux autres syndromes, dont ceux d'Aicardi [24], de Catel-Manzke, de Noonan [25], du nævus basocellaire, de Meckel-Gruber, de Walker-Warburg, de Rubinstein-Taybi, de Pai, de Goldenhar, de Wolf-Hirschhorn ou syndrome 4p; la microphtalmie de type Lenz; le syndrome MIDAS (microphthalmia, dermal aplasia, and sclerocornea); l'hypoplasie dermique focale de Goltz; les trisomies 13 et 18. Enfin, de très nombreuses associations polymalformatives, non syndromiques, ont aussi été décrites. Outre un examen clinique pédiatrique complet, nous recommandons la pratique systématique d'examens complémentaires afin de mettre en évidence ces syndromes ou associations polymalformatifs (Tableau 21-4) [26,27].
Trois grands types de complications sont à connaître : le décollement de rétine (DR), les néovaisseaux; la cataracte. L'incidence cumulée du DR n'est pas connue; certains auteurs ont avancé des chiffres allant jusqu'à 40 % . Il convient de proscrire les sports à risque de traumatisme oculaire et de prescrire des lunettes avec des montures en plastique, afin de limiter au maximum les chocs. Le risque de décollement dépendrait principalement de la profondeur du colobome (donc en fait de l'angle entre la rétine normale et le fond du colobome), bien mesurée en échographie B [28]. Le décollement survient le plus souvent à partir d'une zone de faiblesse sur cette ligne de jonction. Son diagnostic précoce repose sur l'éducation des parents puis de l'enfant et la pratique d'une surveillance puis d'une autosurveillance avec test à l'écran hebdomadaire. Cette éducation est à ce jour insuffisante et la majorité des DR est diagnostiquée trop tardivement, à un stade où il n'existe plus d'indication chirurgicale. L'intérêt du laser argon en prophylaxie du décollement demeure controversé [29]; il est peu ou pas pratiqué en France en 2017. Le diagnostic d'un DR récent sur colobome requiert une analyse clinique et paraclinique – tomographie par cohérence optique (optical coherence tomography [OCT]) et/ou échographie B – précise, afin de déterminer la nature du traitement requis [30]. Plusieurs cas de résorption spontanée de formes limitées à la macula en lien avec des colobomes papillaires ont été rapportés [31]; la situation s'apparente alors sans doute à celle rencontrée en association avec les fossettes colobomateuses (voir plus loin). Les DR récents compliquant les colobomes choriorétiniens constituent généralement une urgence chirurgicale. Le traitement fait appel à une vitrectomie par la pars plana, suivie d'un tamponnement par gaz ou huile de silicone [32-34]. Les complications néovasculaires des colobomes uvéaux sont plus rares mais peuvent survenir chez l'enfant; les anti-vascular endothelial growth factor (anti-VEGF) sont désormais utilisés dans le cadre de leur prise en charge, avec un effet parfois prolongé [35, 36]. La cataracte advient précocement chez les enfants ou jeunes adultes atteints de colobome : cataracte localisée à la zone de défect irien dans le cas d'un colobome irien, et/ou cataracte corticonucléaire classique [37]. Sa prise en charge chirurgicale comporte un risque augmenté de complications rétiniennes et l'indication repose sur la gêne fonctionnelle qui doit être évaluée avec un soin particulier.
Alors que la définition des colobomes repose sur un mécanisme physiopathologique assuré et connu de longue date, la définition de la papille en fleur de liseron est strictement clinique : il s'agit d'une papille de grande taille, d'où émanent de trop nombreux vaisseaux en rayon de roue semblant partir de la périphérie de la papille; un matériel glial recouvre l'excavation papillaire; un liseré de pigmentation pathologique entoure la papille; l'anomalie est généralement unilatérale et la papille est parfois animée de contractions battant sur un rythme propre [38]; dans environ un quart des cas, elle est associée à des signes de persistance de la vascularisation fœtale de type persistance de l'artère hyaloïde (fig. 21-10) [39].

Tableau 21-4 Bilan recommandé devant un colobome uvéal (irien, choriorétinien ou papillaire).
La papille en fleur de liseron a longtemps été considérée comme une forme particulière de colobome papillaire. Cette hypothèse est aujourd'hui très improbable et cette anomalie est considérée comme résultant d'une insuffisance de fermeture de la partie la plus antérieure du pédicule optique et non de la partie postérieure de la fente embryonnaire; il s'agirait donc d'une dysgénésie mésodermique. Plusieurs auteurs ont émis l'hypothèse qu'il s'agirait d'une forme particulière, très postérieure et limitée, de persistance de la vascularisation fœtale [39-41].
La papille en fleur de liseron s'intègre parfois dans un syndrome ou une association polymalformative. L'association papille en fleur de liseron, encéphalocèle transsphénoïdale, et dans sa forme complète, macrocrânie, nez aplati, hypertélorisme modéré, encoche médiane de la lèvre supérieure et fente médiane du palais mou [42] doit être suspectée devant la dysmorphie faciale caractéristique; elle sera confirmée par l'imagerie cérébrale. Le syndrome PHACE (Posterior fossa malformations, Hemangiomas, Arterial anomalies, Cardiac defects and coarctation of the Aorta, Eye abnormalities, and Sternal abnormalities or ventral developmental defects) ou syndrome neurocutané de Pascual-Castroviejo de type II, peut comporter une papille en fleur de liseron. Plus souvent, seulement certaines malformations de ce syndrome sont présentes, certains ont parlé à ce propos de « spectre » PHACE. Enfin, des anomalies de la vascularisation cérébrale de type hypoplasie de l'artère carotide interne ipsilatérale ou de ses branches sont très fréquemment retrouvées. Elles sont compatibles avec des hypoplasies congénitales à l'imagerie, cependant pour certains auteurs, elles seraient l'expression d'une vasculopathie évolutive, avec un risque de moya moya, justifiant d'une surveillance systématique par des IRM cérébrales régulières [43].
Le bilan à la recherche de malformations associées comportera donc essentiellement, outre un examen clinique pédiatrique et dermatologique, une IRM encéphalique comportant des séquences vasculaires.
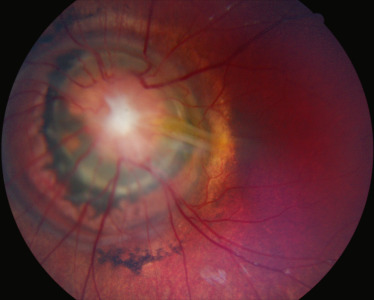
Fig. 21-10 Papille en fleur de liseron typique.
Grande taille, nombreux vaisseaux en rayon de roue semblant partir de la périphérie de la papille ; matériel glial recouvrant l’excavation papillaire ; liseré de pigmentation pathologique entourant la papille ; persistance de l’artère hyaloïde.
Le terme « fossette colobomateuse » , spécifiquement français (optic disc pit en anglais), témoin d'un passé où toutes les malformations du nerf optique étaient considérées comme des « colobomes » , est un faux ami : ces fossettes ne sont aucunement des variantes de colobome; elles n'en partagent aucune des caractéristiques.
On désigne par « fossette colobomateuse » une dépression de forme ronde ou ovalaire, de couleur grise voire jaunâtre, située à l'intérieur de la papille optique, généralement dans le secteur temporal (fig. 21-11). La malformation est généralement unilatérale (dans 85 % des cas), s'accompagne de l'émergence en son sein d'une ou de deux artères ciliorétiniennes dans la moitié des cas et n'affecte le plus souvent pas la vision, d'où une découverte généralement fortuite. Dans certains cas, un déficit campimétrique relié à la tache de Mariotte peut être associé. Histologiquement, une fossette colobomateuse correspond à une hernie de rétine dysplasique au sein d'une bourse de tissu collagène atteignant l'espace sous-arachnoïdien en arrière de la lame criblée. Les fossettes colobomateuses sont des malformations isolées et leur découverte ne requiert donc aucun bilan particulier. Le mécanisme est probablement génétique (gène inconnu), avec une transmission semblant dominante autosomique [44].
Le problème posé par les « fossettes colobomateuses » de la papille est la maculopathie qui les complique dans un pourcentage inconnu des cas (25 à 75 % des cas selon les études), le plus souvent chez l'adulte, plus rarement à l'adolescence, exceptionnellement chez le jeune enfant. Celle-ci début généralement par un schisis de la rétine interne, n'affectant le plus souvent pas ou très peu la fonction visuelle et ne requérant alors aucun traitement, ce d'autant que sa résorption spontanée est possible (fig. 21-12). Dans un second temps, un décollement séreux de l'épithélium pigmentaire peut venir compliquer le schisis initial; une altération durable de la fonction visuelle peut en résulter et constitue alors une indication opératoire. En présence d'une maculopathie sans indication de traitement, on recommandera un suivi clinique semestriel assorti d'une autosurveillance avec pour instruction de consulter dans l'intervalle en cas d'altération de la fonction visuelle. Le mécanisme de ces maculopathies est matière à débat. Dans un premier temps, le liquide proviendrait du vitré liquéfié ou/et du liquide cérébrospinal, avec un épuisement des fonctions de pompage de l'épithélium pigmentaire à certaines périodes correspondant aux phases de décompensation; dans un second temps, une composante véritablement rhegmatogène viendrait compliquer le processus. Le traitement chirurgical spécifique de ces DR particuliers combine généralement photocoagulation au laser péripapillaire et vitrectomie par la pars plana avec tamponnement par gaz [45].
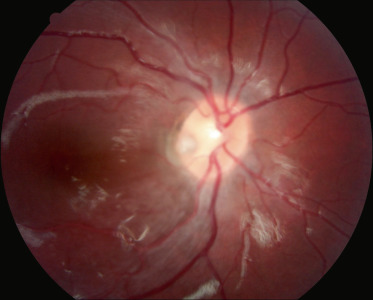
Fig. 21-11 Fossette colobomateuse de la papille.
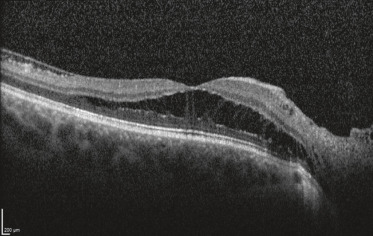
Fig. 21-12 Schisis de la rétine interne compliquant une fossette colobomateuse de la papille, avec préservation de la fonction visuelle.
Il s'agit là encore d'un faux ami, premièrement parce que ce syndrome a longtemps été désigné sous le nom de « syndrome rein-colobome » , alors que la malformation papillaire décrite dans nombre de cas n'est pas colobomateuse; deuxièmement parce qu'un authentique colobome papillaire ou choriorétinien peut s'associer à des malformations rénales, constituant alors un authentique syndrome « rein-colobome » au sens strict du terme.
Par « syndrome papillo-rénal » on désigne désormais l'association d'une malformation spécifique de la papille optique à une hypoplasie rénale généralement bilatérale. Cette malformation consiste en une excavation centrée de la papille (donc non colobomateuse) et une absence (ou un estompement) de vaisseaux rétiniens; à leur place on note la présence de nombreux vaisseaux ciliorétiniens émanant de l'anneau neurorétinien [46].
La transmission est génétique, dominante autosomique; le rôle des mutations du gène PAX2, autrefois incriminé dans environ la moitié des cas de syndrome « rein-colobome » , n'est pas clair.
L'importance de reconnaître ce syndrome réside dans le dépistage et la prise en charge précoce de l'insuffisance rénale qui résulte des malformations rénales associées (échographie rénale de départ et surveillance régulière de la pression artérielle, de l'ionogramme sanguin, urée, créatininémie, bandelette urinaire).
Cette expression désigne certes une malformation de la papille, qui « attire l'attention » du clinicien, mais celle-ci est l'un des éléments d'un syndrome plus complet et affectant l'ensemble du pôle postérieur, où l'hémirétine supérotemporale (située au-dessus d'une ligne passant par la papille et la fovéa) est normale avec une hémipapille surélevée, tandis que l'hémirétine inféronasale est ectasique, myopique, avec une choroïdose myopique évolutive réalisant un staphylome inférieur et une hémipapille étalée postérieurement (fig. 21-13). L'émergence des vaisseaux rétiniens est inversée, en temporal de la papille. La ligne de démarcation entre la rétine normale et la rétine myopique détermine le grand axe de la papille ainsi que l'axe positif de l'astigmatisme myopique associé qui peut faire suspecter le syndrome [47]. Le syndrome est bilatéral, non héréditaire; sa physiopathologie est méconnue et son histoire naturelle peu claire en raison d'un manque de description chez le nourrisson. Il s'accompagne d'anomalies campimétriques dans les zones du champ visuel correspondant aux défects en cellules ganglionnaires [48]. La réalisation d'un champ visuel chez ces patients est importante : des cas de scotome hémianopsique bitemporal voire d'hémianopsie bitemporale ont été rapportés, en rapport avec des tumeurs suprasellaires [5]. En cas d'anomalie campimétrique douteuse, une imagerie est donc indiquée.
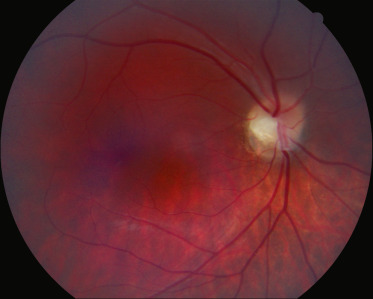
Fig. 21-13 Syndrome de dysversion papillaire.
Papille optique dysverse ; situs inversus partiel de l’émergence des vaisseaux rétiniens ; surélévation de l’hémipapille supérieure ; croissant scléral inférieur ; staphylome rétinien inférieur.
Les complications sont l'apanage de l'adolescent et surtout de l'adulte : décollements séreux rétiniens secondaires aux anomalies de jonction entre les rétines normale et ectasique; néovascularisation choroïdienne au bord inférieur du staphylome; drusen de la papille optique ou congestions veineuses rétiniennes résultant du tassement papillaire.
Généralement traitées avec les malformations du nerf optique, elles ne le concernent cependant pas directement. Celui-ci peut-être normal ou variablement malformé.
Les lacunes choriorétiniennes sont des zones rondes ou ovalaires, bien limitées, avec absence de rétine et de choroïde (fig. 21-14). Elles sont le signe d'une dysplasie de la choriorétine, dont elles partagent la valeur d'orientation. Il n'est pas surprenant que dans de nombreux cas, elles s'associent sur le même œil ou l'œil controlatéral à des signes plus sévères de dysplasie choriorétinienne pouvant aller à l'extrême jusqu'à la présence d'un pli falciforme.
Leur localisation péripapillaire chez une fille (ou un garçon atteint de syndrome de Klinefelter) est assez spécifique du syndrome d'Aicardi, qui leur associe des spasmes infantiles avec hypsarrythmie à l'électroencéphalogramme, ainsi qu'une agénésie du corps calleux (triade classique). Il existe souvent aussi une microcéphalie, une polymicrogyrie, des hétérotopies périventriculaires, des malformations vertébrales et une dysmorphie faciale. Le nerf optique – et le reste de l'œil – est rarement complètement normal. De très nombreuses malformations associées ont été rapportées (microphtalmies, colobomes iriens et choriorétiniens, hypoplasies papillaires, pigmentation congénitale du nerf optique). Le gène responsable, encore inconnu, est situé sur le chromosome X. Tous les cas sont sporadiques; la présence de la mutation chez les garçons serait létale in utero [24].
Les dysplasies choriorétiniennes (et donc les lacunes choriorétiniennes qui en sont l'une des possibles expressions) ne sont cependant pas spécifiques du syndrome d'Aicardi : elles s'associent à des formes de microcéphalie avec ou sans lymphœdème des membres inférieurs, avec ou sans déficit intellectuel, de transmission autosomique, récessive ou dominante, avec alors souvent mutations dans le gène KIF11 [49, 50]. Leur localisation est alors variable.
Normalement, le nerf optique en avant de la lame criblée (la papille optique) n'est pas myélinisé et ne contient donc pas d'oligodendrocytes. Chez environ 0,5 % de la population, ce n'est pas le cas : des oligodendrocytes sont présents en avant de la lame criblée et produisent des gaines de myéline autour de certains axones; on parle alors de « fibres à myéline » . Celles-ci apparaissent sous la forme de stries blanchâtres, localisées préférentiellement le long des faisceaux temporaux supérieur et inférieur, dès la papille (fig. 21-15); dans d'autres cas (20 % ), un plumeau périphérique séparé de la papille est observé. Parfois enfin, les fibres à myéline sont étendues à la totalité du pôle postérieur à l'exception de la fovéa et de la périphérie (fig. 21-16); elles entrent alors dans le cadre du syndrome fibres à myéline-myopie-amblyopie [51]. Dans tous les cas, l'anomalie peut être uni- ou plus rarement (20 % ) bilatérale. Elle est parfois héréditaire avec un mode de transmission dominant autosomique.
L'association syndromique, exceptionnelle, au syndrome de Gorlin (nævomatose basocellulaire, de transmission dominante autosomique) est cependant à connaître : la dysmorphie faciale parfois discrète, les malformations du squelette, les kystes de la mâchoire et l'hyperkératose palmoplantaire, qui confère une sensation de granité à la palpation, précèdent généralement l'apparition des nævi, mais pas celle des médulloblastomes (5 à 10 % des patients), cause de décès d'autant plus précoce que le diagnostic est tardif [52].
Les fibres à myéline ont longtemps été considérées comme n'étant que des malformations (des anomalies congénitales et non évolutives). Elles le sont en effet bien souvent; cependant toute lésion de la lame criblée (traumatisme, œdème papillaire de stase, drusen papillaires, etc.) peut aussi être suivie de l'apparition, de l'évolution, parfois de la disparition de fibres à myéline [53-55].
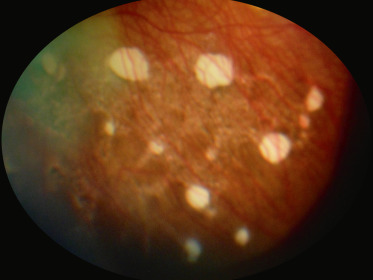
Fig. 21-14 Lacunes choriorétiniennes.
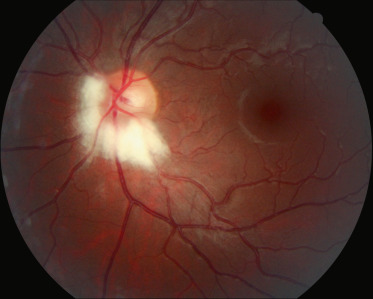
Fig. 21-15 Plumeau de fibres à myéline partant de la papille optique.
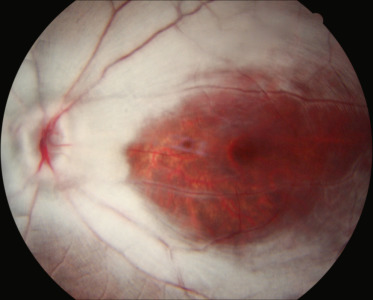
Fig. 21-16 Syndrome fibres à myéline-myopie-amblyopie.
Il s'agit d'une malformation caractérisée par une excavation profonde du fond d'œil centrée par la papille, qui siège au fond de l'excavation. La papille est de morphologie normale (notamment la vascularisation rétinienne est normale) et présente parfois une pâleur temporale. L'anomalie est généralement unilatérale. Elle ne doit pas être confondue avec une papille en fleur de liseron, elle aussi parfois située au fond d'une excavation parfois considérable [56]. L'anomalie est généralement isolée. Exceptionnellement, elle s'inscrirait dans un cadre syndromique (encéphalocèle transsphénoïdale, syndrome PHACE, syndrome du nævus sébacé, syndrome 18q-), justifiant de la réalisation systématique d'une IRM encéphalique avec séquences vasculaires [4]. La physiopathologie n'est pas claire, elle fait probablement intervenir une différenciation sclérale incomplète après la fin du développement de la papille optique, vers le 5 mois de grossesse.
La présence anormale de mélanine en avant de la lame criblée confère aux papilles une teinte vraiment grise. En soi, l'anomalie n'affecte pas la vision, mais elle entre parfois dans un cadre syndromique plus large et peut s'associer à d'autres anomalies (hypoplasie papillaire) responsables d'une mauvaise fonction visuelle [57].
Il faut distinguer ces pigmentations congénitales vraies des pigmentations péripapillaires associées à certaines hypoplasies papillaires, mais aussi des colorations grisâtres temporaires rapportées par Beauvieux dans certains retards de maturation et de la teinte parfois grisâtre des papilles hypoplasiques des albinismes oculocutanés, résultant très probablement d'un effet de contraste avec un fond d'œil hypopigmenté [4].
Pour parler d'aplasie « du nerf optique » , dont l'aplasie « papillaire » fait partie, celui-là doit être complètement absent : absence de fibres ganglionnaires, absence de vaisseaux rétiniens (ou au maximum présence de rares vaisseaux rétiniens rudimentaires). Si une papille est reconnaissable, aussi petite soit-elle, on parle d'hypoplasie papillaire. Dans l'aplasie papillaire, le lieu du nerf optique peut apparaître blanchâtre, mais sans structure identifiable. On distingue des formes unilatérales, généralement isolées chez des enfants par ailleurs en bonne santé et des formes bilatérales, entrant dans le cadre de syndromes variés affectant le système nerveux central [5, 58].
Il existe alors deux papilles optiques. L'anomalie est généralement unilatérale et la vision de l'œil atteint est rarement normale. Le plus souvent, l'une d'entre elle correspond à un colobome choriorétinien inférieur prenant l'aspect d'une papille anormale; on a alors une papille principale et une papille satellite inférieure [59, 60]. L'anomalie peut aussi n'être pas colobomateuse. L'IRM peut montrer deux nerfs optiques juxtaposés.
Certaines malformations du nerf optique n'entrent dans aucune des catégories décrites dans ce chapitre. On les range donc dans cette catégorie à part, qui par définition est extrêmement hétérogène. Dans son acception contemporaine restreinte, la dysplasie inclassable n'est donc ni un colobome, ni une fleur de liseron, ni une papille de syndrome papillo-rénal, ni un staphylome péripapillaire. Une association à des encéphalocèles transsphénoïdales a été rapportée [42]. Nous recommandons la pratique d'une imagerie cérébrale systématique dans de tels cas.
Nous avons volontairement souhaité traiter la prise en charge des malformations papillaires dans leur ensemble. Elles ont longtemps été synonymes du renoncement de l'ophtalmopédiatre à l'obtention d'une vision utile. De ce passé demeurent des croyances et des habitudes profondément ancrées dans les pratiques.
Leur prise en charge doit être dictée par le pragmatisme et le bon sens en évitant l'acharnement inutile et dommageable et, surtout, le renoncement prématuré à une rééducation efficace de la part fonctionnelle d'une amblyopie organique. Trois principes doivent dicter l'attitude au cas par cas du clinicien :
- – l'avenir fonctionnel d'un œil non amblyope n'est jamais garanti et l'amblyothérapie de ce jour est la prévention de la cécité pour les années à venir. Cet avenir est d'autant moins garanti que l'œil non amblyope est lui aussi malformé (par exemple porteur d'un colobome n'affectant pas l'acuité mais à risque de DR);
- – les plus sévères des malformations papillaires (vastes colobomes papillaires ou choriorétiniens, papilles en fleur de liseron, fibres à myéline extensives) peuvent être compatibles avec une vision utile, c'est-à-dire chiffrable;
- – il n'existe pas de corrélation robuste entre l'aspect anatomique d'une malformation papillaire et le potentiel de vision de l'œil atteint.
Par principe, nous recommandons donc un essai « systématique » de rééducation de la part fonctionnelle d'une amblyopie organique liée à une malformation papillaire, et ce dès l'étape du diagnostic. Cette rééducation se distingue de celle d'une amblyopie fonctionnelle : elle ne peut en effet consister qu'en une occlusion séquentielle, et ce jusqu'aux 10 ans de l'enfant. Elle est donc beaucoup plus lourde et contraignante qu'une occlusion d'amblyopie fonctionnelle. Elle consiste en quelques minutes d'occlusion par jour assorties d'une stimulation visuelle chez le nouveau-né. Si après plusieurs semaines d'occlusion séquentielle quotidienne dans le cas d'une malformation sévère, le comportement de l'enfant demeure celui d'un enfant aveugle (le nourrisson généralement pleure avant de s'endormir; il n'existe pas de réponse aux stimulations visuelles), alors l'occlusion peut être arrêtée. Il est probable que certaines amblyothérapies chez le nouveau-né et le nourrisson, même arrêtées plus tard, permettent de prévenir l'apparition d'un syndrome du monophtalme fonctionnel congénital. Il est certain qu'en cas de nystagmus manifeste latent, meilleure est l'acuité de l'œil amblyope, meilleure sera la vision binoculaire finale. Il est également certain qu'une vision utile sur un œil amblyope est un atout précieux sur le long terme.
[1] Borchert M, Garcia-Filion P The syndrome of optic nerve hypoplasia Curr Neurosci Rep: ( 2008 ) :8: 395-403
[2] Zeki SM, Dudgeon J, Dutton GN Reappraisal of the ratio of disc to macula/disc diameter in optic nerve hypoplasia Br J Ophthalmol: ( 1991 ) :75: 538-541
[3] Jonas JB, Gusek GC, Naumann GO Optic disc, cup and neuroretinal rim size, configuration and correlations in normal eyes Invest Ophthalmol Vis Sci: ( 1988 ) :29: 1151-1158
[4] Brodsky MC :-
[5] Garcia-Filion P, Borchert M Prenatal determinants of optic nerve hypoplasia : review of suggested correlates and future focus Surv Ophthalmol: ( 2013 ) :58: 610-619
[6] Oatman OJ, McClellan DR, Olson ML, Garcia-Filion P Endocrine and pubertal disturbances in optic nerve hypoplasia, from infancy to adolescence Int J Pediatr Endocrinol: ( 2015 ) :2015: 8-
[7] Rivkees SA, Fink C, Nelson M, Borchert M Prevalence and risk factors for disrupted circadian rhythmicity in children with optic nerve hypoplasia Br J Ophthalmol: ( 2010 ) :94: 1358-1362
[8] Jutley-Neilson J, Harris G, Kirk J The identification and measurement of autistic features in children with septo-optic dysplasia, optic nerve hypoplasia and isolated hypopituitarism Res Dev Disabil: ( 2013 ) :34: 4310-4318
[9] Taylor D Congenital tumours of the anterior visual system with dysplasia of the optic discs Br J Ophthalmol: ( 1982 ) :66: 455-463
[10] Fink C, Garcia-Filion P, Borchert M Failure of stem cell therapy to improve visual acuity in children with optic nerve hypoplasia J AAPOS: ( 2013 ) :17: 490-493
[11] Jacobson L, Hellstrom A, Flodmark O Large cups in normal-sized optic discs : a variant of optic nerve hypoplasia in children with periventricular leukomalacia Archives of Ophthalmology: ( 1997 ) :115: 1263-1269
[12] Lennartsson F, Nilsson M, Flodmark O, Jacobson L Damage to the immature optic radiation causes severe reduction of the retinal nerve fiber layer, resulting in predictable visual field defects Invest Ophthalmol Vis Sci: ( 2014 ) :55: 8278-8288
[13] Landau K, Bajka JD, Kirchschlager BM Topless optic disks in children of mothers with type I diabetes mellitus Am J Ophthalmol: ( 1998 ) :125: 605-611
[14] Takagi M, Abe H, Hatase T Superior segmental optic nerve hypoplasia in youth Jpn J Ophthalmol: ( 2008 ) :52: 468-474
[15] Novakovic P, Taylor DS, Hoyt WF Localising patterns of optic nerve hypoplasia-retina to occipital lobe Br J Ophthalmol: ( 1988 ) :72: 176-182
[16] Hoyt WF, Rios-Montenegro EN, Behrens MM, Eckelhoff RJ Homonymous hemioptic hypoplasia. Fundoscopic features in standard and red-free illumination in three patients with congenital hemiplegia Br J Ophthalmol: ( 1972 ) :56: 537-545
[17] Mann I :-
[18] Gopal L, Badrinath SS, Kumar KS Optic disc in fundus coloboma Ophthalmology: ( 1996 ) :103: 2120-2126 discussion 6-7
[19] Tellier AL, Cormier-Daire V, Abadie V CHARGE syndrome : report of 47 cases and review Am J Med Genet: ( 1998 ) :76: 402-409
[20] Blake KD, Davenport SL, Hall BD CHARGE association : an update and review for the primary pediatrician Clin Pediatr (Phila): ( 1998 ) :37: 159-173
[21] Verloes A Updated diagnostic criteria for CHARGE syndrome : a proposal Aam J Med Genet A: ( 2005 ) :133: 306-308
[22] Berends MJ, Tan-Sindhunata G, Leegte B, van Essen AJ Phenotypic variability of Cat-Eye syndrome Genet Couns: ( 2001 ) :12: 23-34
[23] Rosias PR, Sijstermans JM, Theunissen PM Phenotypic variability of the cat eye syndrome. Case report and review of the literature Genet Couns: ( 2001 ) :12: 273-282
[24] Aicardi J Aicardi syndrome Brain Dev: ( 2005 ) :27: 164-171
[25] Lee NB, Kelly L, Sharland M Ocular manifestations of Noonan syndrome Eye (London, England): ( 1992 ) :6: 328-334
[26] Denis D, Girard N, Levy-Mozziconacci A Ocular coloboma and results of brain MRI: preliminary results Journal Francais d'Ophtalmologie: ( 2013 ) :36: 210-220
[27] Huynh N, Blain D, Glaser T Systemic diagnostic testing in patients with apparently isolated uveal coloboma Am J Ophthalmol: ( 2013 ) :156: 1159-1168 e4
[28] Venincasa VD, Modi YS, Aziz HA Clinical and echographic features of retinochoroidal and optic nerve colobomas Invest Ophthalmol Vis Sci: ( 2015 ) :56: 3615-3620
[29] Uhumwangho OM, Jalali S Chorioretinal coloboma in a paediatric population Eye (London, England): ( 2014 ) :28: 728-733
[30] Gopal L, Khan B, Jain S, Prakash VS A clinical and optical coherence tomography study of the margins of choroidal colobomas Ophthalmology: ( 2007 ) :114: 571-580
[31] Baumann SL, Sadeghi Y, Wolfensberger TJ Cyclical spontaneous resorption of serous macular detachment associated with optic disc coloboma Klin Monbl Augenheilkd: ( 2016 ) :233: 522-523
[32] Liu T, Zhang M, Xu B Evaluation of the safety of sclerotomy incision in patients with choroidal colobomas with/without associated microcornea Retina: ( 2014 ) :34: 2300-2305
[33] Ramezani A, Dehghan MH, Rostami A Outcomes of retinal detachment surgery in eyes with chorioretinal coloboma Journal of Ophthalmic & Vision Research: ( 2010 ) :5: 240-245
[34] Chang S, Gregory-Roberts E, Chen R Retinal detachment associated with optic disc colobomas and morning glory syndrome Eye (London, England): ( 2012 ) :26: 494-500
[35] Naithani P, Vashisht N, Mandal S Intravitreal bevacizumab in choroidal neovascularization associated with congenital choroidal and optic nerve coloboma in children : long-term improvement in visual acuity J AAPOS: ( 2010 ) :14: 288-290
[36] Bhende M, Suganeswari G, Gopal L Choroidal neovascularization associated with coloboma of the choroid : a series of three cases Indian J Ophthalmol: ( 2011 ) :59: 148-151
[37] Mohamed A, Chaurasia S, Ramappa M Lenticular changes in congenital iridolenticular choroidal coloboma Am J Ophthalmol: ( 2014 ) :158: 827-830 e2
[38] Kindler P Morning glory syndrome: unusual congenital optic disk anomaly Am J Ophthalmol: ( 1970 ) :69: 376-384
[39] Fei P, Zhang Q, Li J, Zhao P Clinical characteristics and treatment of 22 eyes of morning glory syndrome associated with persistent hyperplastic primary vitreous Br J Ophthalmol: ( 2013 ) :97: 1262-1267
[40] Brown GC, Gonder J, Levin A Persistence of the primary vitreous in association with the morning glory disc anomaly J Pediatr Ophthalmol Strabismus: ( 1984 ) :21: 5-7
[41] Parsa CF, Robert MP Thromboembolism and congenital malformations : from Duane syndrome to thalidomide embryopathy JAMA Ophthalmol: ( 2013 ) :131: 439-447
[42] Brodsky MC, Hoyt WF, Hoyt CS Atypical retinochoroidal coloboma in patients with dysplastic optic discs and transsphenoidal encephalocele Archives of Ophthalmology: ( 1995 ) :113: 624-628
[43] Traboulsi EI Morning glory disk anomaly–more than meets the eye J AAPOS: ( 2009 ) :13: 333-334
[44] Georgalas I, Ladas I, Georgopoulos G, Petrou P Optic disc pit : a review Graefes Arch Clin Exp Ophthalmol: ( 2011 ) :249: 1113-1122
[45] Jain N, Johnson MW Pathogenesis and treatment of maculopathy associated with cavitary optic disc anomalies Am J Ophthalmol: ( 2014 ) :158: 423-435
[46] Parsa CF, Silva ED, Sundin OH Redefining papillorenal syndrome: an underdiagnosed cause of ocular and renal morbidity Ophthalmology: ( 2001 ) :108: 738-749
[47] Shinohara K, Moriyama M, Shimada N Analyses of shape of eyes and structure of optic nerves in eyes with tilted disc syndrome by swept-source optical coherence tomography and three-dimensional magnetic resonance imaging Eye (London, England): ( 2013 ) :27: 1233-1241 quiz 42
[48] Pichi F, Romano S, Villani E Spectral-domain optical coherence tomography findings in pediatric tilted disc syndrome Graefes Arch Clin Exp Ophthalmol: ( 2014 ) :252: 1661-1667
[49] Alzial C, Dufier JL, Aicardi J Ocular abnormalities of true microcephaly Ophthalmologica: ( 1980 ) :180: 333-339
[50] Balikova I, Robson AG, Holder GE Ocular manifestations of microcephaly with or without chorioretinopathy, lymphedema or intellectual disability (MCLID) syndrome associated with mutations in KIF11 Acta Ophthalmol: ( 2016 ) :94: 92-98
[51] Tarabishy AB, Alexandrou TJ, Traboulsi EI Syndrome of myelinated retinal nerve fibers, myopia, and amblyopia : a review Surv Ophthalmol: ( 2007 ) :52: 588-596
[52] De Jong PT, Bistervels B, Cosgrove J Medullated nerve fibers. A sign of multiple basal cell nevi (Gorlin's) syndrome Arch Ophthalmol: ( 1985 ) :103: 1833-1836
[53] Shah M, Park HJ, Gohari AR, Bhatti MT Loss of myelinated retinal nerve fibers from chronic papilledema J Neuroophthalmol: ( 2008 ) :28: 219-221
[54] Duval R, Hammamji K, Aroichane M Acquired myelinated nerve fibers in association with optic disk drusen J AAPOS: ( 2010 ) :14: 544-547
[55] Prakalapakorn SG, Buckley EG Acquired bilateral myelinated retinal nerve fibers after unilateral optic nerve sheath fenestration in a child with idiopathic intracranial hypertension J AAPOS: ( 2012 ) :16: 534-538
[56] Kim SH, Choi MY, Yu YS, Huh JW Peripapillary staphyloma : clinical features and visual outcome in 19 cases Arch Ophthalmol: ( 2005 ) :123: 1371-1376
[57] Brodsky MC, Buckley EG, McConkie-Rosell A The case of the gray optic disc! Surv Ophthalmol: ( 1989 ) :33: 367-372
[58] Weiter JJ, McLean IW, Zimmerman LE Aplasia of the optic nerve and disk Am J Ophthalmol: ( 1977 ) :83: 569-576
[59] Donoso LA, Magargal LE, Eiferman RA, Meyer D Ocular anomalies simulating double optic discs Can J Ophthalmol: ( 1981 ) :16: 85-87
[60] Vedantham V Double optic discs, optic disc coloboma, and pit : spectrum of hybrid disc anomalies in a single eye Arch Ophthalmol: ( 2005 ) :123: 1450-1452
P. Lebranchu
Un gonflement de la papille optique peut être secondaire à un œdème des fibres axonales. L'origine de cet œdème papillaire peut être liée à une inflammation locale (uvéite intermédiaire, neurorétinite, etc., voir chapitre 14), une inflammation du nerf optique (névrite optique), une ischémie des axones (neuropathie vasculaire, rarissime en pédiatrie) ou une accumulation du liquide céphalorachidien au niveau des gaines des nerfs optiques (hypertension intracrânienne, compression). Beaucoup plus fréquemment, la papille adopte un aspect gonflé qui n'est pas secondaire à un œdème des axones. Les faux œdèmes papillaires peuvent en effet se rencontrer en cas de petite papille de l'hypermétrope, d'infiltration papillaire (rare), de malformation congénitale (persistance de tissu glial prépapillaire) ou des drusen de la papille optique (fréquents). Dans une étude prospective récente rapportant le diagnostic final d'enfants adressés en ophtalmopédiatrie pour œdème papillaire de stase, ce diagnostic n'était confirmé que dans 2 cas sur 34 (26 présentaient un faux œdème, les autres un œdème inflammatoire ou sans diagnostic de certitude). Les maux de tête étaient présents dans 26 cas sur 34, sans lien évident avec un vrai ou un faux œdème [1]. La démarche diagnostique face à une surélévation de la papille de l'enfant sera donc essentielle, car certaines étiologies sont totalement bénignes alors que d'autres sont potentiellement mortelles.
Les drusen de la papille optique correspondent à des dépôts hyalins, calciques et acellulaires apparaissant dans une papille pleine. Cette anomalie serait secondaire à une lame criblée de petite taille, qui ralentirait le flux axoplasmique et provoquerait une souffrance axonale. La libération du matériel mitochondrial lors de la dégénérescence axonale serait à l'origine de dépôts qui se calcifient avec le temps, et qui aggravent progressivement la souffrance des axones. Ces dépôts initialement profonds tendent à migrer vers la surface du nerf optique chez l'adulte jeune [2].
Les drusen restent longtemps asymptomatiques, particulièrement chez l'enfant. Leur découverte est donc en général fortuite lors de la réalisation d'un fond d'œil systématique. L'acuité visuelle est en général conservée, une baisse d'acuité visuelle n'étant présente que dans 5 % des cas. Chez l'adulte, les troubles du champ visuel sont présents chez environ 50 % des patients [3, 4], plus souvent en cas de visibilité de drusen. Les déficits les plus fréquents sont arciformes (inférieur plus souvent que supérieur) et un élargissement de la tache aveugle, aboutissant parfois à un rétrécissement concentrique du champ visuel. Ces altérations sont concordantes avec la perte en fibres mesurée par OCT en cas de visibilité des drusen [4]. Cependant des troubles du champ visuel peuvent être déjà présents et sévères chez l'enfant [5]. Les complications sont rares, mais il faudra évoquer un néovaisseau parapapillaire en cas de baisse brutale de la vision. Les drusen papillaires sont bilatéraux dans 66 à 85 % des cas. Ils se rencontrent chez 0,4 % des enfants (prépondérance féminine), mais jusqu'à 2 % des adultes caucasiens [2]. Des formes familiales ont été décrites (transmission autosomique dominante) ainsi que des associations syndromiques (pseudoxanthome élastique, dystrophies rétiniennes, syndromes d'Alagille ou de Joubert) [5].
Le diagnostic est aisé lorsqu'ils sont superficiels, apparaissant comme des petits dépôts ronds, beiges et réfringents. Cependant, chez le sujet jeune, ils se localisent profondément dans la papille, provoquant une surélévation papillaire qui peut mimer un œdème papillaire (fig. 21-17). La distinction entre œdème et drusen est alors ténue, mais essentielle sur le plan diagnostique. Les drusen ont une évolution lente et majoritairement bénigne, alors qu'un œdème papillaire bilatéral requiert un diagnostic urgent en raison de ses étiologies potentiellement mortelles. Des particularités cliniques orientent vers un diagnostic de drusen, car la papille apparaît avec un bord festonné, sans hyperhémie ni masquage des vaisseaux superficiels. Une vascularisation rétinienne anormale est parfois associée : ramification précoce, vaisseaux surnuméraires, tortuosité vasculaire (cependant certaines de ces anomalies sont également observables en cas d'œdème papillaire [6]). La présence de pulsation veineuse spontanée oriente alors le diagnostic vers un faux œdème car le pouls veineux disparaît en cas d'hypertension intracrânienne. Aucun traitement n'a actuellement fait la preuve de son efficacité. Un traitement hypotonisant est recommandé en cas de drusen provoquant un déficit évolutif du champ visuel.
Certains examens complémentaires confirment le diagnostic de drusen. L'échographie en mode B pourrait être le meilleur examen paraclinique, objectivant la surélévation papillaire associée à du matériel hyperéchogène lorsque le signal échographique est abaissé (fig. 21-18). L'autofluorescence spontanée de la papille optique n'est pas systématique, mais sa présence oriente fortement le diagnostic (fig. 21-19). Une étude sur 32 yeux de drusen confirmés par échographie mode B chez l'enfant (7 à 17 ans) objectivait une autofluorescence spontanée dans 92 % des drusen profonds et 100 % des drusen superficiels [7]. Le scanner peut mettre en évidence des drusen profonds, lorsque ceux-ci sont calcifiés (fig. 21-20). Cependant ces trois examens (échographie, autofluorescence et scanner) ne sont positifs qu'après calcification des drusen. Le problème diagnostique reste donc entier en cas de drusen profonds non calcifiés, situation courante en pédiatrie.
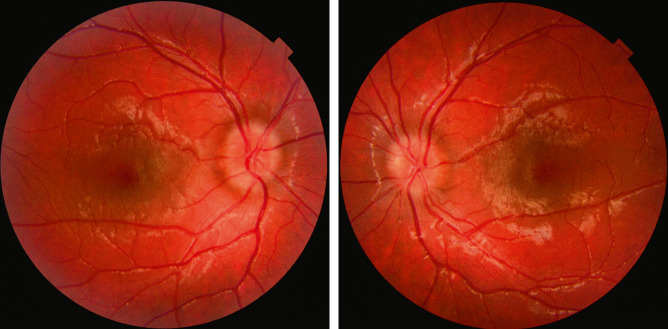
Fig. 21-17 Drusen papillaires profonds bilatéraux chez une jeune fille de 9 ans, sans antécédents, adressés pour bilan de céphalées.
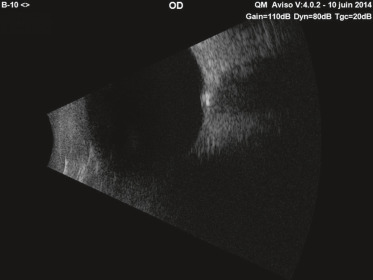
Fig. 21-18 Échographie en mode B du globe oculaire (drusen profonds). Même patiente qu’à la figure 21-17.
Noter l’hyperéchogénicité spontanée de la tête du nerf optique, signe de drusen calcifiés.
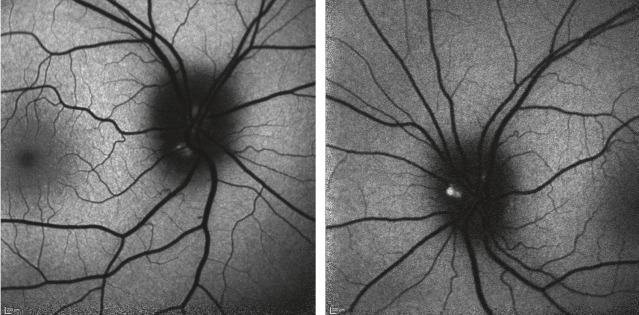
Fig. 21-19 Autofluorescence spontanée (drusen profonds). Même patiente qu’à la figure 21-17.
Noter les zones d’hyper-autofluorescence spontanée dans le nerf optique, correspondant aux drusen.
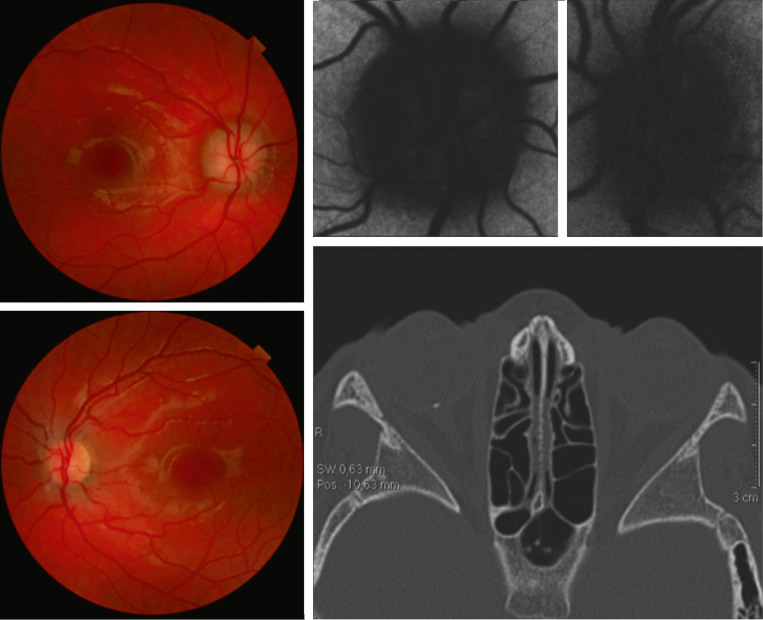
Fig. 21-20 Garçon 9 ans, adressé en urgence pour découverte fortuite d’un oedème papillaire droit sans baisse d’acuité visuelle.
Autofluorescence considérée comme normale (a posteriori, une hypofluorescence centrale peut se deviner). Le scanner révèle une calcification de la tête du nerf optique droit, confirmant le diagnostic de drusen.
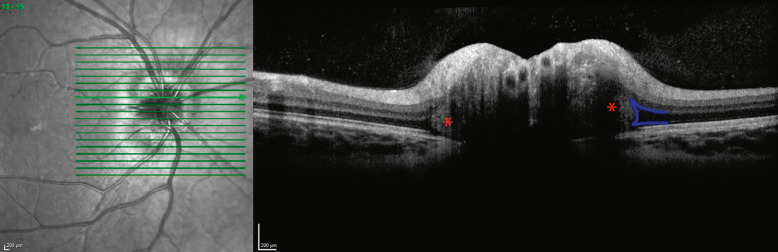
Fig. 21-21 Coupe OCT passant par la papille droite (drusen profonds). Même patiente qu’à la figure 21-17.
Noter l’accumulation de matériel hyperréflective avec un bord clairement individualisable situé de part et d’autre à la sortie du canal scléral (étoile rouge). Cette accumulation de matériel écarte l’EPR de la CNE, provoquant un élargissement de l’espace hyporéflectif (signe de la botte, trait bleu).
Pour résoudre ce problème, plusieurs équipes ont proposé d'utiliser l'OCT pour visualiser directement les drusen profonds dans le canal scléral (fig. 21-21). La littérature n'est pas encore claire sur l'aspect en OCT. Certains retrouvent une masse hyperréflective avec un bord clairement individualisable [8] située à la sortie du canal scléral – entre l'épithélium pigmenté rétinien (EPR) et la couche nucléaire externe (CNE) –, soulevant la couche des fibres ganglionnaires. L'espace hyporéflectif formé entre l'EPR et la CNE prend alors la forme d'une botte (signe de la botte). D'autres équipes proposent d'utiliser l'enhanced depth imaging-optical coherence tomography (EDI-OCT) pour objectiver les drusen profonds [9], retrouvant de petites bandes courtes hyperréflectives (drusen en formation?) entourant parfois une région ronde hyporéflective (drusen formé?). Cependant, la pertinence du diagnostic de drusen sur l'analyse qualitative de l'OCT reste faible, comprise entre 50 et 64 % [10]. En particulier, certains cas d'œdème papillaire chronique sont également associés à la présence de matériel hyperréflectif sous la couche des fibres ganglionnaires, au pourtour de la papille optique, dont l'origine n'est actuellement pas claire.
D'autres équipes ont proposé d'utiliser le mode retinal nerve fiber layer (RNFL) de l'OCT pour distinguer des drusen d'un œdème papillaire en fonction de l'épaisseur de la couche des fibres ganglionnaires. L'adulte présente souvent une atrophie du RNFL, corrélée au déficit campimétrique [4]. Les enfants (fig. 21-22) présentent en général des drusen profonds, associés significativement à une augmentation globale de l'épaisseur du RNFL par rapport au groupe contrôle [5]. Cette épaisseur est également majorée en cas d'œdème papillaire, et la simple lecture du RNFL ne permet pas de distinguer clairement drusen et œdème [11]. L'angiographie à la fluorescéine garde alors sa place chez l'enfant lorsque tous les examens sont négatifs, retrouvant en cas de drusen profonds une coloration nodulaire précoce (25 % ) ou tardive (29 % ) du disque, une coloration péripapillaire circonférentielle (80 % ) mais en aucun cas une diffusion du colorant en dehors du disque [12]. En cas de doute, une surveillance est recommandée et les mesures par OCT (qualitatives et quantitatives) deviennent alors des éléments utiles de suivi, avec élévation stable (non variable) du RNFL dans les mois qui suivent.
L’essentiel
Les drusen papillaires de l’enfant sont souvent découverts fortuitement. L’acuité visuelle est conservée, le champ visuel (lorsqu’il est réalisable) peut déjà montrer un déficit. Ils sont en général profonds et bilatéraux, et tout le problème sera de les différencier d’un oedème papillaire bilatéral. Certains examens complémentaires peuvent permettre le diagnostic (l’échographie en mode B à une meilleure sensibilité que l’autofluorescence). La place de l’OCT (coupe du nerf optique en mode EDI, RNFL) est débattue. Au moindre doute, un scanner ou une IRM doivent être réalisés. Devant une baisse profonde de la vision, il faudra évoquer une complication néovasculaire.
[1] Kovarik JJ, Doshi PN, Collinge JE, Plager DA Outcome of pediatric patients referred for papilledema J AAPOS: ( 2015 ) :19: 344-348
[2] Auw-Haedrich C, Staubach F, Witschel H Optic disk drusen Surv Ophthalmol: ( 2002 ) :47: 515-532
[3] Wilkins JM, Pomeranz HD Visual manifestations of visible and buried optic disc drusen J Neuroophthalmol: ( 2004 ) :24: 125-129
[4] Gili P, Flores-Rodríguez P, Martin-Ríos MD, Carrasco Font C Anatomical and functional impairment of the nerve fiber layer in patients with optic nerve head drusen Graefes Arch Clin Exp Ophthalmol: ( 2013 ) :251: 2421-2428
[5] Noval S, Visa J, Contreras I Visual field defects due to optic disk drusen in children Graefes Arch Clin Exp Ophthalmol: ( 2013 ) :251: 2445-2450
[6] Pilat AV, Proudlock FA, McLean RJ Morphology of retinal vessels in patients with optic nerve head drusen and optic disc edema Invest Ophthalmol Vis Sci: ( 2014 ) :55: 3484-3490
[7] Gili P, Flores-Rodríguez P, Yangüela J, Herreros Fernández ML Using autofluorescence to detect optic nerve head drusen in children J AAPOS: ( 2013 ) :17: 568-571
[8] Lee KM, Woo SJ, Hwang JM Differentiation of optic nerve head drusen and optic disc edema with spectral-domain optical coherence tomography Ophthalmology: ( 2011 ) :118: 971-977
[9] Merchant KY, Su D, Park SC Enhanced depth imaging optical coherence tomography of optic nerve head drusen Ophthalmology: ( 2013 ) :120: 1409-1414
[10] Kulkarni KM, Pasol J, Rosa PR, Lam BL Differentiating mild papilledema and buried optic nerve head drusen using spectral domain optical coherence tomography Ophthalmology: ( 2014 ) :121: 959-963
[11] Karam EZ, Hedges TR Optical coherence tomography of the retinal nerve fibre layer in mild papilloedema and pseudopapilloedema Br J Ophthalmol: ( 2005 ) :89: 294-298
[12] Pineles SL, Arnold AC Fluorescein angiographic identification of optic disc drusen with and without optic disc edema J Neuroophthalmol: ( 2012 ) :32: 17-22
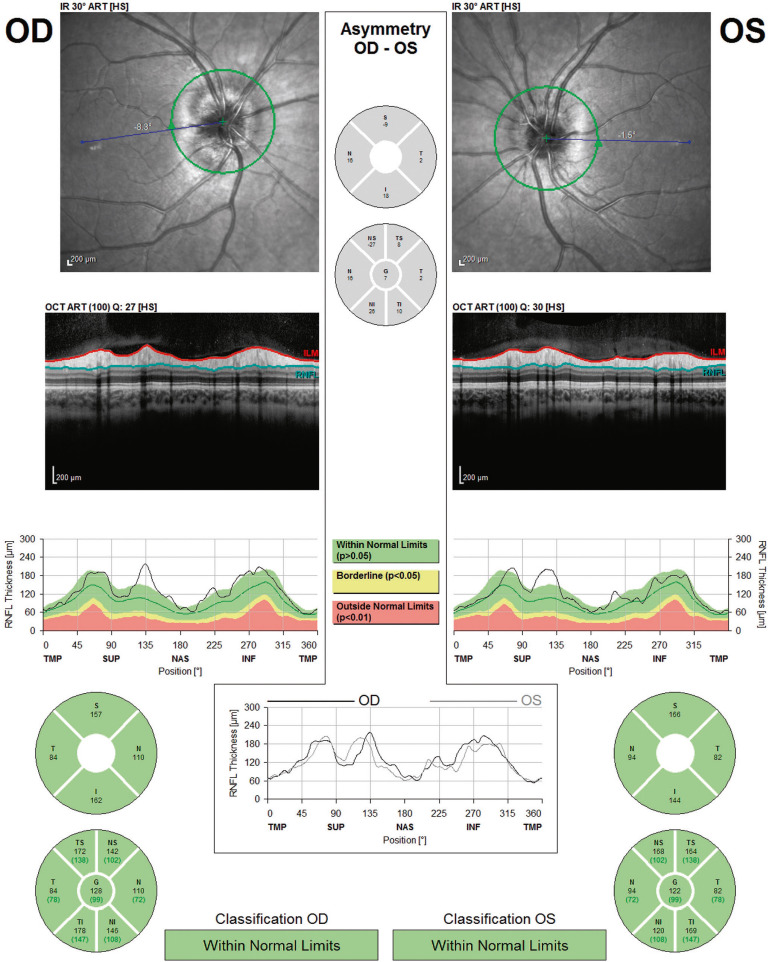
Fig. 21-22 Mesure (RNFL, Heidelberg) de l'épaisseur de la couche des fibres ganglionnaires (drusen profonds). Même patiente qu'à la figure 21-17.
Noter l’épaisseur du nerf optique à la limite supérieur de la normale.
P. Lebranchu
L’incidence annuelle de la neuropathie optique inflammatoire est de 5/100 000 habitants, survenant dans 5 % des cas chez les enfants [1]. Elle se manifeste par une baisse d’acuité visuelle subaiguë, fréquemment associée à une douleur augmentée par la mobilisation du globe [2]. Elle peut survenir dans les suites immédiates d’une infection ou d’une vaccination, avec une fièvre rapportée dans les 15 jours précédents chez 66 % des patients [3]. Elle est deux fois plus fréquente chez les filles, survenant dans 33 à 42 % des cas de façon bilatérale [4, 5]. La baisse d’acuité visuelle initiale est en général très profonde, inférieure à 1/10 dans 69 % des cas [5]. Cependant le pronostic est favorable, l’acuité visuelle finale étant supérieure à 5/10 dans plus de 80 à 90 % des cas [6]. Toutefois certaines formes peuvent avoir un pronostic extrêmement défavorable en l’absence de traitement précoce (voir plus loin « Cas particulier de la neuromyélite optique de Devic » ). Ainsi un traitement (voir plus loin) doit être systématiquement proposé en cas de baisse visuelle profonde, avec une surveillance rapprochée. Une seconde ligne thérapeutique sera également discutée en cas de non-récupération après 2 à 3 semaines. Le déficit du champ visuel n’est pas toujours systématisé (fig. 21-23), objectivant souvent un scotome cæcocentral ou un élargissement de la tache aveugle [2]. L’examen du fond d’oeil peut être normal ou observer un oedème papillaire : sa fréquence est double par rapport à la névrite optique de l’adulte, une papillite étant observée dans 67 % des cas.
Le bilan biologique comprendra des recherches sérologiques (syphilis, Lyme, griffe du chat, virus neurotropes, etc.), inflammatoires (sarcoïdose, etc.) et auto-immunes (anticorps antinucléaires, etc.). Une neuro-imagerie (fig. 21-24) est systématique et urgente, retrouvant des anomalies dans près de un cas sur deux [5]. Elle sera éventuellement complétée par une analyse du liquide céphalorachidien selon l’avis des neuropédiatres (mesure de la pression d’ouverture, recherche d’une synthèse intrathécale d’immunoglobuline, dosage des lactates, etc.). L’examen de neuro-imagerie permettra souvent d’orienter le diagnostic et de renseigner sur le pronostic. Dans une étude rétrospective de 26 cas [4], la moitié était des névrites optiques isolées, dont la grande majorité présentait une IRM initiale normale. Parmi les 12 patients présentant des anomalies neurologiques, la moitié était des encéphalomyélites aiguës disséminées, cinq autres ont évolué vers une sclérose en plaques (Tableau 21-5).
La neuromyélite optique de Devic reste un diagnostic rare, représentant 3 à 4 % des névrites de l’adolescent [4, 5]. Son pronostic sans traitement est extrêmement péjoratif sur le plan visuel (cécité légale) mais également sur le plan général (conséquence de la myélite). Son identification précoce est absolument nécessaire, car un traitement précoce peut permettre à la fois une récupération et un contrôle des poussées ultérieures. Dans une étude rétrospective de 9 cas [7], la moyenne d’âge des patients lors de l’attaque initiale était de 14 ans. La névrite optique était bilatérale (d’emblée pour 5 cas, séquentielle pour 2 cas), entraînant une baisse visuelle extrêmement profonde chez plus de la moitié des patients. La myélite transverse précédait ou accompagnait la poussée de névrite optique dans 6 cas sur 9. Le dosage sanguin des anticorps anti-neuromyélite optique (anti-NMO) était positif dans 7 cas sur 9. La recherche sanguine des anticorps anti-NMO et anti-myelin oligodendrocyte glycoprotein (anti-MOG) doit donc être réalisée systématiquement en cas de suspicion de maladie de Devic : neuropathie optique bilatérale sévère ou neuropathie optique unilatérale ne répondant que partiellement à la corticothérapie.
Comme chez l’adulte, il existe après une névrite optique un risque non négligeable d’évolution vers une sclérose en plaques (fig. 21-25). Celui-ci est estimé globalement entre 19 et 36 % [5, 8], passant à 6 ans de 7 % en cas d’IRM initiale normale à 42 % en cas d’IRM anormale [4]. Cependant chez l’adolescent, la névrite optique est le mode d’entrée dans la maladie inflammatoire démyélinisante dans moins de 20 % des cas [9]. Les facteurs protecteurs sont une névrite optique cliniquement isolée, un âge de survenue précoce (avant 10 ans), un antécédent d’infection dans les 15 jours précédents ou le sexe masculin. Au contraire, la présence, sur l’imagerie initiale, d’un hypersignal encéphalique (en dehors de celui situé sur le nerf optique) [5], une synthèse intrathécale d’immunoglobuline [8] ou la récurrence de la névrite optique [10] sont des facteurs de risque forts d’évolution vers une sclérose en plaques. L’influence du caractère bilatéral de la névrite optique est actuellement débattue, une étude l’identifiant comme protecteur [8], une autre comme facteur de risque [5]. L’ensemble de ces facteurs, associé aux données cliniques, permettra d’orienter le traitement et la surveillance de cette névrite optique.
En cas de suspicion de névrite optique démyélinisante, une corticothérapie intraveineuse de 20 à 30 mg/kg/j sera proposée (sans dépasser la dose de 1 g/j) pendant 3 à 5 jours éventuellement poursuivie par un relai per os de 1 mg/kg/j diminué sur 14 à 21 jours. En aucun cas, une corticothérapie per os d’emblée ne sera proposée (elle augmenterait le risque d’entrée dans la maladie et la fréquence des poussées chez l’adulte). En l’absence de réponse à 21 jours, il faut savoir évoquer des formes sévères de névrite optique et en particulier la neuromyélite optique de Devic. Une seconde ligne thérapeutique, avec de nouvelles perfusions de corticoïdes, des perfusions intraveineuses d’immunoglobuline [9] ou des échanges plasmatiques doit être discutée.
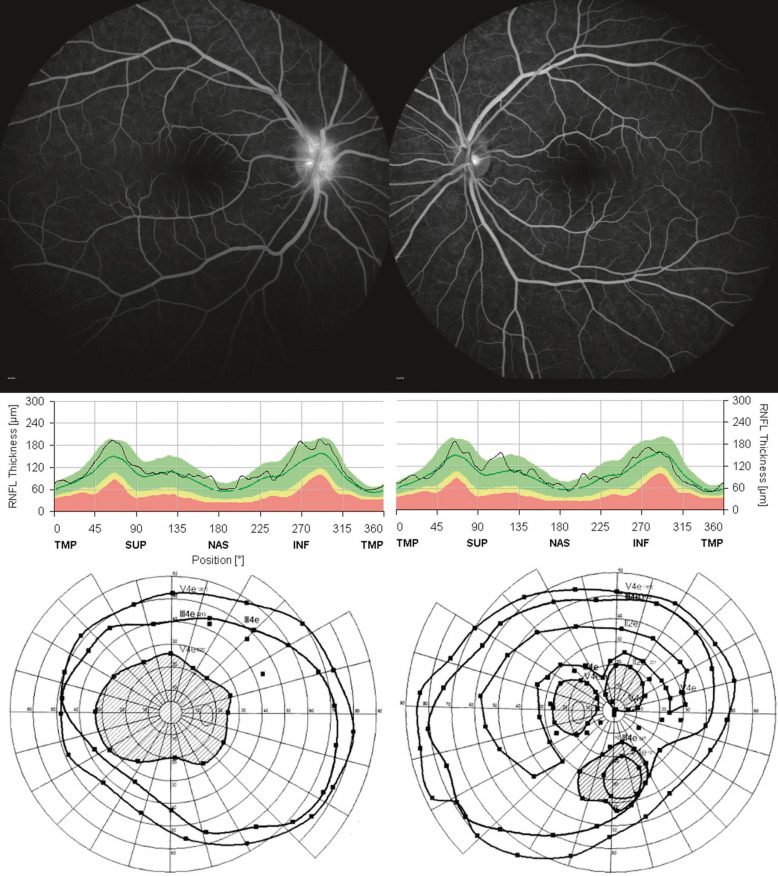
Fig. 21-23 Jeune fille 17 ans présentant des douleurs rétro-orbitaires depuis 15 jours.
Baisse d’acuité visuelle bilatérale depuis 48 heures (perception de la lumière à droite ; 0,14 P14 à gauche) ; pas d’anomalie du segment antérieur ; très légère hyperhémie papillaire droite, sans anomalie à gauche ; l’angiographie confirme la diffusion du colorant à droite (en haut à gauche), sans oedème papillaire franc (RFNL normal, au centre) ; scotome central important à droite, atteinte paracentrale gauche. Les PEV confirment l’atteinte bilatérale du nerf optique ; l’IRM initiale est considérée comme normale. La patiente est hospitalisée pour 5 jours de bolus de corticoïdes, puis par échange plasmatique 1 mois plus tard en raison d’une récupération sous-optimale. Les anticorps anti-MOG se sont avérés positifs, faisant porter le diagnostic de maladie du spectre NMO.

Tableau. 21-5 Neuropathie optique inflammatoire de l’enfant.
ADEM : acute demyelinating encephalomyelitis ; CRP : C-reactive protein ; CMV : cytomégalovirus ; EBV : virus d’Epstein-Barr ; HSV : herpes simplex virus ; IRM : imagerie par résonance magnétique ; MOG : myelin oligodendrocyte glycoprotein ; NFS : numération formule sanguine ; NMO : neuromyélite optique ; SEP : sclérose en plaques ; VZV : virus zona-varicelle.
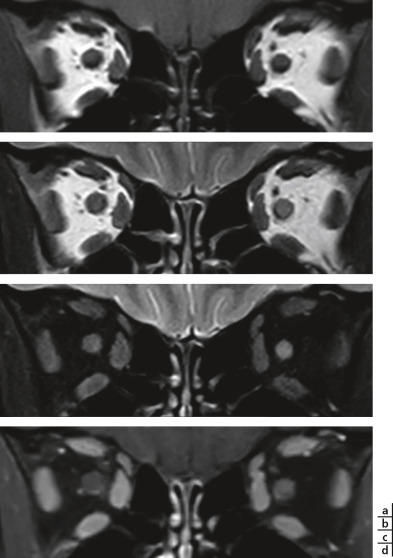
Fig. 21-24 Névrite optique gauche dans le cadre d’une sclérose en plaques. IRM orbitaire 3 teslas, coupes coronales.
a. Sur la séquence pondérée en T1, aucune différence n’apparaît entre les deux nerfs optiques. b, c. Le nerf optique gauche apparaît en hypersignal par rapport au droit sur la séquence pondérée en T2 (b), et cela apparaît encore plus visible sur cette même séquence avec saturation de la graisse (c). d. Enfin l’injection de gadolinium provoque un rehaussement du nerf gauche par rapport au droit sur la séquence T1 avec saturation de graisse. (Remerciements au Dr A.S. Delemazure.)
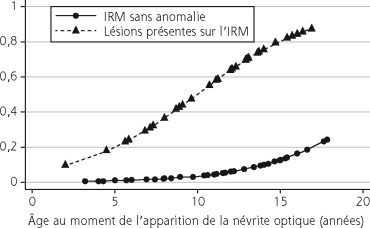
Fig. 21-25 Probabilité de développer une sclérose en plaques chez l’enfant, en fonction de l’âge de survenue de la neuropathie optique (en abscisse) et de la présence (courbe avec les triangles) ou de l’absence (courbe avec les points) d’anomalie visible lors de l’IRM initiale (une ou plusieurs anomalies de signal pondérées T2 en dehors des voies visuelles).
Méta-analyse regroupant 223 patients. (D’après Waldman AT et al. Pediatric optic neuritis and risk of multiple sclerosis : Meta-analysis of observational studies. J AAPOS 2011 ; 15 : 441-6.)
[1] Rodriguez M, Siva A, Cross SA Optic neuritis : a population-based study in Olmsted County, Minnesota Neurology: ( 1995 ) :45: 244-250
[2] Roussat B, Gohier P, Doummar D Acute optic neuritis in children : clinical features and treatment. A study of 28 eyes in 20 children J Fr Ophtalmol: ( 2001 ) :24: 36-44
[3] Boomer JA, Siatkowski RM Optic neuritis in adults and children Semin Ophthalmol: ( 2003 ) :18: 174-180
[4] Alper G, Wang L Demyelinating optic neuritis in children J Child Neurol: ( 2009 ) :24: 45-48
[5] Wilejto M, Shroff M, Buncic JR The clinical features, MRI findings, and outcome of optic neuritis in children Neurology: ( 2006 ) :67: 258-262
[6] Wan MJ, Adebona O, Benson LA Visual outcomes in pediatric optic neuritis Am J Ophthalmol: ( 2014 ) :158: 503-507 e2
[7] Lotze TE, Northrop JL, Hutton GJ Spectrum of pediatric neuromyelitis optica Pediatrics: ( 2008 ) :122: e1039-e1047
[8] Lucchinetti CF, Kiers L, O'Duffy A Risk factors for developing multiple sclerosis after childhood optic neuritis Neurology: ( 1997 ) :49: 1413-1418
[9] Dale RC, Brilot F, Banwell B Pediatric central nervous system inflammatory demyelination : acute disseminated encephalomyelitis, clinically isolated syndromes, neuromyelitis optica, and multiple sclerosis Curr Opin Neurol: ( 2009 ) :22: 233-240
[10] Absoud M, Cummins C, Desai N Childhood optic neuritis clinical features and outcome Arch Dis Child: ( 2011 ) :96: 860-862
P. Lebranchu
Tout œdème papillaire bilatéral de l'enfant doit faire réaliser une neuro-imagerie en urgence, car la cause la plus fréquente d'hypertension intracrânienne (HTIC) de l'enfant est dominée par la pathologie tumorale (voir chapitre 20), éventuellement infectieuse (méningite virale, etc.). Rarement un bilan initial négatif fera évoquer une HTIC idiopathique pédiatrique. L'HTIC idiopathique correspond à une élévation de la pression intracrânienne en absence d'hydrocéphalie, de processus expansif intracrânien, ou d'inflammation du liquide céphalorachidien. Aucune étude n'a évalué l'incidence de l'HTIC idiopathique avant 18 ans. Celle de l'adulte est environ de 1/100 000. Son diagnostic clinique repose sur un ensemble de critères nécessaires, récemment mis à jour [1] :
- – observation d'un œdème papillaire de stase bilatéral (parfois asymétrique, rarement unilatéral) ou d'une paralysie oculomotrice du VI;
- – associée à un examen neurologique normal;
- – associée à une neuro-imagerie comportant des séquences vasculaires veineuses excluant les causes secondaires d'HTIC;
- – associée à une composition normale du liquide céphalorachidien (LCR);
- – associée à une pression d'ouverture du LCR supérieure à 28 cm d'HO (ou 25 cm d'HO en l'absence de sédation).
L'IRM permet d'éliminer les causes secondaires radiologiques d'HTIC, dont la thrombose des sinus duraux de la base du crâne. Elle permet également d'objectiver des signes indirects d'HTIC, comparables à ceux de l'adulte [2] : aplatissement du pôle postérieur des globes oculaires, élargissement de la gaine de nerfs optiques et selle turcique vide (fig. 21-26). L'interrogatoire et le bilan biologique doivent éliminer d'autres causes d'HTIC [1], en particulier les intoxications médicamenteuses (tétracycline, rétinoïdes, vitamine A, lithium, thyroxine, hormone de croissance, etc.) et les problèmes médicaux (maladie d'Addison, hypoparathyroïdisme, anémie, insuffisance rénale chronique, syndrome de Turner, trisomie 21, etc.). Le déficit en vitamine D pourrait représenter jusqu'à 25 % des causes d'HTIC secondaire de l'enfant [3]. Parmi les mineurs présentant une HTIC idiopathique, 40 % ont moins de 10 ans [4].
La prépondérance féminine augmente avec l'âge, passant d'un patient sur deux avant 10 ans à plus de 90 % après 18 ans. La prévalence de l'obésité augmente également avec l'âge, un surpoids étant présent chez 43 % des enfants de moins de 10 ans mais chez presque 90 % des 15-17 ans [5]. La pression d'ouverture du liquide céphalorachidien pourrait pour certains varier avec l'âge, avec une norme inférieure à : 82 mm d'HO chez les nouveau-nés, 180 mm d'HO avant 8 ans, 200 mm d'HO avant 18 ans et 250 mm d'HO après [4]. Pour d'autres, la pression d'ouverture du LCR ne varie pas en fonction de l'âge de l'enfant, et est considérée comme anormale (> 90 percentile) au-delà de 28 cm d'HO [6].
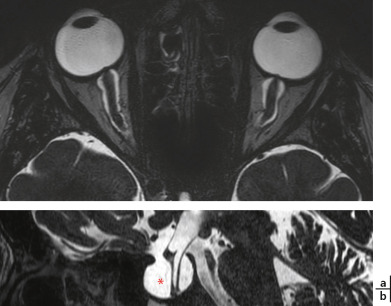
Fig. 21-26 Signes indirects d’HTIC à l’IRM.
a. T2 3D : dilatation des gaines du nerf optique, aplatissement du pôle postérieur des globes oculaires. Noter l’oedème papillaire visualisable comme une protrusion sombre à l’émergence du nerf optique dans le globe oculaire. b. Selle turcique vide (astérisque rouge), visualisée sur une reconstruction sagittale en IRM (séquence T2). Noter la tige pituitaire qui plonge dans la selle turcique, et l’absence d’hypophyse visualisable.
(Remerciements au Dr A.S. Delemazure.)
Sa présentation clinique diffère peu de celle de l'adulte, le patient se plaignant fréquemment de céphalées, d'éclipses visuelles, d'acouphènes pulsatiles et de raideur de nuque. La céphalée est typiquement exacerbée par les manœuvres positionnelles ou de Valsalva. Contrairement à l'adulte, l'atteinte de la sixième paire crânienne est relativement fréquente, pouvant atteindre jusqu'à 80 % des patients avant l'âge de 11 ans [7]. Au moment du diagnostic, 90 % des enfants présentent un déficit du champ visuel et 20 % une baisse d'acuité visuelle. La très grande majorité des enfants présentent un œdème lors de leur consultation (fig. 21-27), qui peut évoluer jusqu'à 33 % vers l'atrophie. La présence simultanée de drusen du nerf optique n'est pas rare, et peut poser un problème de diagnostic différentiel. Une étude a retrouvé une association entre drusen et œdème chez 43 % des enfants [8]. Cependant cette étude peut être critiquée car le diagnostic a été retenu sur l'image OCT du nerf optique, dont la valeur diagnostique entre drusen et œdème est actuellement extrêmement faible (voir chapitre 21.2). Une atteinte permanente de l'acuité visuelle peut survenir chez 10 % des patients, et environ 20 % présentent une altération persistante de leur champ de vision [8, 9]. Les séquelles visuelles définitives sont significativement liées à la présence d'emblée d'un important œdème papillaire (grade de Frisen> 3), évoluant à la fois vers une perte en fibres au niveau du nerf optique (13 % ) associée parfois à une altération des lignes des photorécepteurs maculaires visible sur l'OCT [8]. Plus que le degré d'acuité visuelle, la surveillance est fondée sur le fond d'œil, le champ visuel et l'OCT (mode RNFL) (Tableau 21-6).
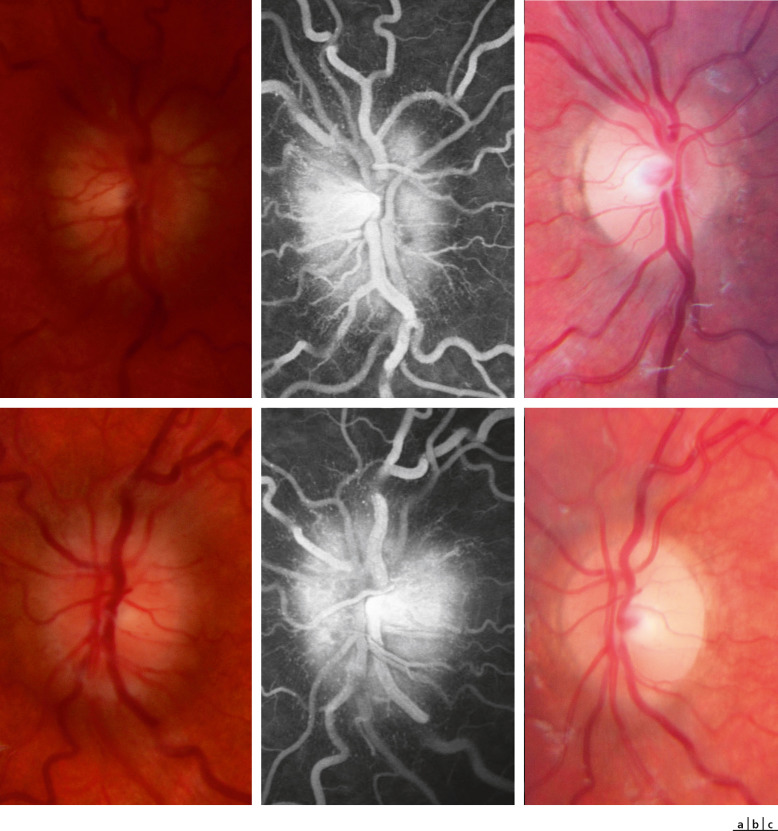
Fig. 21-27 OEdème papillaire bilatéral (grade 3 de Frisen) chez un enfant de 8 ans présentant depuis 3 semaines des céphalées en casque et des vomissements matinaux (oeil droit en haut, oeil gauche en bas).
a. Aspect de l’oedème papillaire bilatéral. Acuité visuelle conservée. b. L’angiographie à la fluorescéine confirme l’oedème isolé. Après bilan, le diagnostic d’HTIC idiopathique est retenu. c. Aspect des papilles après 3 mois de traitement par acétazolamide per os. L’HTIC clinique récidivera 1 mois après l’arrêt des traitements.
Tableau 21-6 – Classification de l’oedème papillaire en fonction des 5 stades de gravité de Frisen.
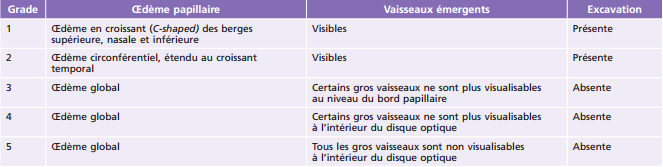
D’après Frisen L. Swelling of the optic nerve head : a staging scheme. J Neurol Neurosurg Psychiatry 1982 ; 45 : 13-8.
La prise en charge de l'HTIC idiopathique de l'enfant et de l'adolescent ne diffère pas de celle de l'adulte, nécessitant une coordination multidisciplinaire. Elle repose sur l'association de mesures hygiéno-diététiques (d'autant plus que l'enfant est âgé), d'une ponction lombaire déplétive et des inhibiteurs de l'anhydrase carbonique per os (15 à 25 mg/kg/j). Le topiramate ou des diurétiques comme le furosémide peuvent être utilisés en seconde ligne de traitement. La place des traitements chirurgicaux est évoquée au cas par cas [4]. La durée du traitement n'est pas standardisée, dépendant de l'évolution de l'œdème et des signes ophtalmologiques. Le pronostic est meilleur chez l'enfant que chez l'adolescent et l'adulte et dans la grande majorité des cas le Diamox peut être arrêté au bout de quelques semaines, en particulier en cas de formes post-virales [10]. Une surveillance ophtalmologique mensuelle est initialement préconisée, modulée en fonction de l'évolution clinique. Le taux de récurrence à l'arrêt du traitement est de 18 % , principalement dans l'année qui suit (Tableau 21-7) [11].
Tableau 21-7 – HTIC chez l’enfant.

HTIC : hypertension intracrânienne ; IRM : imagerie par résonance magnétique ; LCR : liquide céphalorachidien, OCT : optical coherence tomography.
[1] Friedman DI, Liu GT, Digre KB Revised diagnostic criteria for the pseudotumor cerebri syndrome in adults and children Neurology: ( 2013 ) :81: 1159-1165
[2] Görkem SB, Doğanay S, Canpolat M MR imaging findings in children with pseudotumor cerebri and comparison with healthy controls Childs Nerv Syst: ( 2015 ) :31: 373-380
[3] Masri A, Jaafar A, Noman R Intracranial hypertension in children : etiologies, clinical features, and outcome J Child Neurol: ( 2015 ) :30: 1562-1568
[4] Standridge SM Idiopathic intracranial hypertension in children : a review and algorithm Pediatr Neurol: ( 2010 ) :43: 377-390
[5] Balcer LJ, Liu GT, Forman S Idiopathic intracranial hypertension : relation of age and obesity in children Neurology: ( 1999 ) :52: 870-872
[6] Avery RA, Shah SS, Licht DJ Reference range for cerebrospinal fluid opening pressure in children N Engl J Med: ( 2010 ) :363: 891-893
[7] Cinciripini GS, Donahue S, Borchert MS Idiopathic intracranial hypertension in prepubertal pediatric patients : characteristics, treatment, and outcome Am J Ophthalmol: ( 1999 ) :127: 178-182
[8] Gospe SM, Bhatti MT, El-Dairi MA Anatomic and visual function outcomes in paediatric idiopathic intracranial hypertension Br J Ophthalmol: ( 2016 ) :100: 505-509
[9] Phillips PH, Repka MX, Lambert SR Pseudotumor cerebri in children J AAPOS: ( 1998 ) :2: 33-38
[10] Ravid S, Shachor-Meyouhas Y, Shahar E Viral-induced intracranial hypertension mimicking pseudotumor cerebri Pediatr Neurol: ( 2013 ) :49: 191-194
[11] Ravid S, Shahar E, Schif A, Yehudian S Visual outcome and recurrence rate in children with idiopathic intracranial hypertension J Child Neurol: ( 2015 ) :30: 1448-1452
P. Lebranchu
Le gliome des voies visuelles représente 90 % des tumeurs du nerf optique de l'enfant, et 1 % des tumeurs intracrâniennes [1], potentiellement responsable d'un œdème du nerf optique. Les gliomes des voies visuelles sont des astrocytomes pilocytiques de bas grade dépendant des voies visuelles afférentes. Ils sont associés dans 33 % des cas à une neurofibromatose de type 1 (NF1) [2], et 13 à 20 % des enfants présentant une NF1 développent un gliome du nerf optique [3, 4]. La recherche de signe de NF1 est donc indispensable, d'autant plus que les gliomes associés à une neurofibromatose ont en général un meilleur pronostic que les gliomes sporadiques [5]. On s'attachera ainsi à rechercher une histoire familiale de NF1, des taches café au lait, des nodules de Lisch, des neurofibromes plexiformes, des lésions osseuses, une pseudo-arthrite, des taches hyperréflectives de Yasunari au fond d'œil, chez l'enfant et ses parents. En cas de NF1, le gliome apparaît en général tôt dans la vie (âge moyen de survenue 5 ans), sans pour autant être symptomatique [1].
Un tiers à la moitié des patients présente une baisse visuelle non douloureuse progressive, associée à une atrophie optique [6]. Il existe un déficit du réflexe pupillaire afférent (DPAR) en cas de pathologie asymétrique. Celle-ci apparaît en général dans l'enfance, mais parfois à l'adolescence. La baisse de vision pourrait être plus fréquente lorsque l'extension de la tumeur se fait en rétrochiasmatique [4]. Les autres paramètres de la vision (champ visuel, sensibilité des contrastes, vision des couleurs) sont également altérés, mais ne sont pas toujours mesurables de façon fiable chez l'enfant. Les potentiels évoqués visuels (PEV) ne sont ni assez sensibles, ni assez spécifiques pour être utilisés couramment en dépistage chez le patient NF1. Une exophtalmie ou un strabisme sont parfois associés. Une extension hypothalamique peut entraîner des conséquences endocriniennes. Le diagnostic est confirmé par l'IRM (lésions hypo- ou iso-intense T1 et hyperintense T2, dont la portion solide se rehausse après injection de gadolinium) (fig. 21-28), classant les gliomes en trois stades selon l'extension :
- – gliome isolé du nerf optique (stade Dodge 1);
- – gliome du chiasma avec ou sans atteinte du nerf (stade Dodge 2);
- – gliome s'étendant vers les structures adjacentes du chiasma comme l'hypothalamus (stade Dodge 3).
Les classifications modifiées permettent un dénombrement plus précis des lésions. En cas d'association à une NF1, une étude histologique n'est pas requise pour le diagnostic (consensus européen); dans les autres cas, une biopsie sera discutée, d'où l'importance de l'examen ophtalmologique de l'enfant et de ses parents [7].
L'histoire naturelle du gliome est variable, la tumeur pouvant rester stable, voire même régresser. En cas de gliome asymptomatique, une attitude observationnelle est recommandée en raison de la croissance lente de la tumeur. Une progression après l'adolescence est rare.
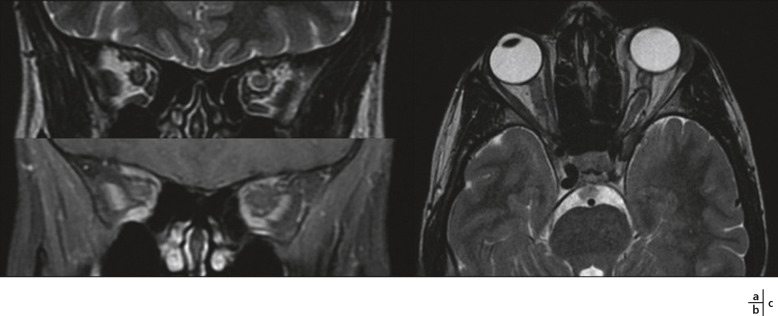
Fig. 21-28 Gliome du nerf optique chez un patient atteint de neurofibromatose de type 1.
a, b. Coupes coronales T2 (a) et T1 avec saturation de graisse et injection de gadolinium. c. Coupe axiale T2. Augmentation bilatérale de la taille du nerf optique prédominant à gauche, apparaissant en hypersignal T2, légèrement rehaussé par l’injection de gadolinium.
(Remerciements au Dr A.S. Delemazure.)
L'ensemble de ces paramètres explique que les modalités de prise en charge des gliomes du nerf optique sont complexes et controversées. Un traitement est proposé aux patients dont le gliome entraîne une dégradation de la fonction visuelle. La chimiothérapie est en général le traitement de première intention, stabilisant l'acuité visuelle chez la majorité des patients [1, 8]. Le traitement chirurgical est réservé à la biopsie en cas de gliomes sans signe de neurofibromatose, ou à la réduction tumorale (debulking) des volumineux gliomes intrinsèques.
Avant 2007, il était recommandé de réaliser un examen ophtalmologique chez tous les enfants atteints de NF1 annuellement jusqu'à l'âge de 6 ans, puis de façon beaucoup plus espacée après 6 ans. Des études récentes ont montré que les gliomes symptomatiques ne sont pas seulement l'apanage des jeunes enfants, mais peuvent progresser au moment de la puberté. Les recommandations européennes préconisent un examen tous les 6 mois jusqu'à 6 ans puis un par an jusqu'à 18 ans [9]. Les recommandations américaines préconisent un examen annuel des patients jusqu'à 8 ans puis tous les 2 ans jusqu'à 18 ans [10]. L'examen ophtalmologique doit alors comprendre une étude des réflexes pupillaires, de l'acuité visuelle, du champ visuel éventuellement couplé à la mesure de la couche des cellules ganglionnaires péripapillaires [11]. L'amincissement de l'épaisseur du nerf optique mesuré en OCT (RNFL) est associé à la perte de vision chez les patients NF1 présentant un gliome [12]. La réalisation systématique d'une IRM ou de PEV est actuellement débattue, mais non recommandée à titre systématique (Tableau 21-8) [1].
Tableau 21-8 – Pathologies tumorales du nerf optique.

DPAR : déficit du reflexe pupillaire afférent ; IRM : imagerie par résonance magnétique ; NF1 : neurofibromatose de type 1 ; NO : nerf optique ; OCT : optical coherence tomography ; PEV : potentiels évoqués visuels ; RNFL : retinal nerve fiber layer ; RPM : réflexe photomoteur.
[1] Nair AG, Pathak RS, Iyer VR, Gandhi RA Optic nerve glioma : an update Int Ophthalmol: ( 2014 ) :34: 999-1005
[2] Janss AJ, Grundy R, Cnaan A Optic pathway and hypothalamic/chiasmatic gliomas in children younger than age 5 years with a 6-year follow-up Cancer: ( 1995 ) :75: 1051-1059
[3] Segal L, Darvish-Zargar M, Dilenge ME Optic pathway gliomas in patients with neurofibromatosis type 1 : follow-up of 44 patients J AAPOS: ( 2010 ) :14: 155-158
[4] Balcer LJ, Liu GT, Heller G Visual loss in children with neurofibromatosis type 1 and optic pathway gliomas: relation to tumor location by magnetic resonance imaging Am J Ophthalmol: ( 2001 ) :131: 442-445
[5] Liu GT Visual loss in childhood Surv Ophthalmol: ( 2001 ) :46: 35-42
[6] Listernick R, Charrow J, Greenwald M, Mets M Natural history of optic pathway tumors in children with neurofibromatosis type 1 : a longitudinal study J Pediatr: ( 1994 ) :125: 63-66
[7] Walker DA, Liu J, Kieran M A multi-disciplinary consensus statement concerning surgical approaches to low-grade, high-grade astrocytomas and diffuse intrinsic pontine gliomas in childhood (CPN Paris 2011) using the Delphi method Neuro-Oncol: ( 2013 ) :15: 462-468
[8] Dodgshun AJ, Elder JE, Hansford JR, Sullivan MJ Long-term visual outcome after chemotherapy for optic pathway glioma in children: Site and age are strongly predictive Cancer: ( 2015 ) :121: 4190-4196
[9] Caen S, Cassiman C, Legius E, Casteels I Comparative study of the ophthalmological examinations in neurofibromatosis type 1. Proposal for a new screening algorithm Eur J Paediatr Neurol: ( 2015 ) :19: 415-422
[10] Listernick R, Ferner RE, Liu GT, Gutmann DH Optic pathway gliomas in neurofibromatosis-1 : controversies and recommendations Ann Neurol: ( 2007 ) :61: 189-198
[11] Chang L, El-Dairi MA, Frempong TA Optical coherence tomography in the evaluation of neurofibromatosis type-1 subjects with optic pathway gliomas J AAPOS: ( 2010 ) :14: 511-517
[12] Avery RA, Cnaan A, Schuman JS Longitudinal change of circumpapillary retinal nerve fiber layer thickness in children with optic pathway gliomas Am J Ophthalmol: ( 2015 ) :160: 944-952
C. Orssaud
Les neuropathies optiques héréditaires (NOH) de l'enfant sont dues à la dégénérescence des cellules ganglionnaires de la rétine (CGR) dont l'axone participe à la constitution des voies optiques prégéniculées (nerf optique, chiasma et tractus optique). Cette dégénérescence porte principalement mais non exclusivement sur les CGR constituant le faisceau maculaire. C'est pourquoi ces NOH sont responsables d'une baisse d'acuité visuelle (BAV) précoce et sévère, due à un scotome central, objectivé lorsque l'âge de l'enfant permet de le rechercher.
Des nombreux gènes de NOH ont été identifiés et les mécanismes physiopathogéniques les sous-tendant sont mieux connus. Citons le rôle des mitochondries qui apparaît comme essentiel [1]. De très nombreux gènes interviennent dans le fonctionnement de cet organite impliqué dans la production énergétique de la cellule ainsi que dans le contrôle de l'équilibre entre fusion et fission des réseaux mitochondriaux intracellulaires. Or cet équilibre est un élément essentiel dans la régulation du stress oxydatif et dans les mécanismes d'apoptose. Ces avancées permettent la mise en place de pistes thérapeutiques pour les années à venir.
Deux classifications des NOH de l'enfant ayant un intérêt clinique peuvent être proposées. Celles-ci ne se superposent pas totalement mais se recoupent et se complètent.
La première de ces classifications, indispensable au conseil génétique, est fondée sur le mode de transmission génétique. Tous les modes peuvent être observés : mitochondrial, dominant autosomique, récessif autosomique et lié au chromosome X. Mais, cette classification a des limites puisque dans une grande série, une mutation d'atrophie optique dominante du gène OPA1 a été retrouvée chez 40 % des patients présentant une NOH sporadique [2]. Des valeurs moins importantes ont été retrouvées dans d'autres études mais qui confirme qu'un certain nombre de cas sporadiques sont en fait dus à des mutations dominantes [3]. Les avancées de la biologie moléculaire ont permis de localiser et d'identifier plusieurs gènes [4] : ainsi il a été possible de préciser au mieux les différentes formes cliniques rencontrées pour chaque mode de transmission.
Il est également intéressant de séparer les NOH de l'enfant selon la présence d'éventuelles manifestations systémiques associées. Cette classification permet de guider les explorations complémentaires à réaliser à la recherche de dysfonctions d'autres organes. Elle ne permet plus de déterminer le gène responsable face à une NOH de l'enfant, puisque certains d'entre eux peuvent donner des formes isolées ou syndromiques.
Lors des formes dites « isolées » , il n'existe qu'un dysfonctionnement du nerf optique. Néanmoins, de rares anomalies extra-oculaires peuvent être retrouvées [1]. C'est pourquoi un bilan systémique est nécessaire. Il peut être orienté par le gène en cause et plus encore par la mutation retrouvée. Le diagnostic des formes isolées ne doit être retenu qu'après avoir éliminé toutes les autres causes de neuropathie optique notamment, chez l'enfant, les causes compressives ou inflammatoires. Cette règle s'applique systématiquement, y compris lors des formes familiales. Le mode de présentation des NOH « syndromiques » est bien différent. L'atteinte du nerf optique n'est que l'un des éléments d'un tableau clinique souvent très riche, associant volontiers des atteintes neurologiques ou métaboliques.
Bien que l'atteinte du nerf optique puisse être la seule manifestation retrouvée, des atteintes extra-oculaires (neurologiques, cardiaques, ORL ou autres), plus ou moins sévères, peuvent et doivent être recherchées [5, 6]. Certaines peuvent nécessiter une prise en charge spécifique. Nous suivrons la répartition des NOH isolées en fonction de leur mode de transmission génétique.
Il faut parler des atrophies optiques dominantes (AOD) car, si le mode de présentation semble assez stéréotypé, l'AOD due à OPA1 (AOD/OPA1) étant considérée comme la forme de référence, de petites différences cliniques pourraient exister en fonction des gènes responsables.
À ce jour, sept gènes ont été rapportés comme étant responsables d'AOD isolées, dont quatre ont été identifiés.
Les gènes OPA1 et OPA3 identifiés depuis plusieurs années, rendent compte que de 60 à 80 % des AOD, mais le premier est le plus fréquemment muté [7]. Ce gène OPA1 localisé sur le chromosome 3 en 3q28-q29, code une dynamine, retrouvée au niveau de la membrane interne des mitochondries, qui permet, en association avec d'autres protéines, leur fusion sous forme de réseaux indispensables à leur bon fonctionnement. Il intervient également dans le contrôle de la réplication de l'acide désoxyribonucléique mitochondrial (ADNmt) et participerait au bon développement de la tête du nerf optique [8]. Les patients porteurs d'une mutation du gène OPA1 auraient une papille dont la surface est statistiquement plus petite que la normale [9]. Enfin, certains polymorphismes de ce gène seraient associés à un risque de survenue de glaucome sans tension [10]. Le gène OPA3 qui s'exprime au sein de la mitochondrie et du peroxyzone, coderait pour une protéine permettant la fission des réseaux mitochondriaux, mécanisme opposé à celui du gène OPA1 Deux autres gènes intervenant également dans le contrôle des réseaux mitochondriaux ont récemment été identifiés : le gène AFG3L2 participant à la fusion des mitochondries, comme OPA1 et le gène DRP1 dans les mécanismes de fission. Leur mutation aboutit à des AOD dont les caractéristiques restent mal définies. Trois autres gènes (OPA4, OPA5 et OPAD8 ) joueraient un rôle dans la survenue d'AOD. Ils ont été localisés mais non identifiés [8].
Initialement appelée maladie de Kjer, cette AOD/OPAJ est la forme la mieux connue parmi les AOD isolées. Elle est appelée dans la littérature « (autosomal) dominant optic atrophy » ou atrophie optique dominante (autosomique). Mais, il semble préférable de préciser le gène muté du fait de l'évolution des connaissances en génétique. Cette NOH ubiquitaire a une prévalence estimée entre 1/25 000 et 1/50 000.
L'AOD/OPAJ présente plusieurs évolutions différentes pouvant coexister au sein d'une même fratrie [1,8 ]. Lors de la forme initialement rapportée, les premières manifestations de cette NOH débutent le plus souvent avant l'âge de 10 ans, toujours avant l'âge de 16 ans. La BAV est symétrique, d'abord minime, pouvant passer longtemps inaperçue. Il est habituel de dire que l'acuité visuelle est proche de 8/10 à l'adolescence. L'évolution est lente sur de nombreuses années, le déficit fonctionnel ne devient gênant qu'aux alentours de l'âge de 40 ou 50 ans [6, 8]. À côté de cette forme d'évolution lente, les progrès de la biologie moléculaire ont permis de rattacher à la AOD/OPA1 une forme plus sévère et précoce au cours desquelles la BAV est brutale, rapidement profonde et « stable » . L'acuité visuelle peut être réduite au décompte des doigts au cours de la seconde décennie ou du moins avant l'âge adulte [1]. Ces formes « précoces » , qui peuvent en imposer pour une NOHL, seraient plus fréquentes chez les garçons que chez les filles. Il faut signaler des formes à début beaucoup plus tardif, au-delà de 50 ans, qui peuvent aider à confirmer le mode de transmission dans les fratries concernées. Toutes les formes peuvent être rencontrées au sein d'une même famille, l'importance de la baisse de vision pouvant être extrêmement variable d'un sujet à l'autre [11].
La principale manifestation de cette est la survenue plus ou moins précoce et rapide d'une BAV selon la forme clinique développée. Plusieurs autres anomalies ophtalmologiques peuvent être retrouvées. Les papilles sont rarement totalement atrophiques. Il existe généralement une pâleur localisée uniquement dans le secteur temporal du fait d'une réduction des capillaires prépapillaires (fig. 21-29). Ces altérations papillaires s'associent à une diminution de volume de la couche des fibres optiques dans tous les quadrants en OCT. Mais cette diminution serait plus marquée dans les secteurs inférieurs [9]. L'excavation papillaire est augmentée chez plus de 50 % des patients porteurs de cette AOD/OPA1. Il est souvent évoqué, mais non totalement prouvé que certains haplotypes du gène OPA1 puissent jouer un rôle favorisant pour le glaucome sans tension [10]. Les nerfs optiques présentent un calibre réduit lors des bilans neuroradiologiques. Si l'existence d'une dyschromatopsie acquise de type bleu-jaune est caractéristique de l'AOD/OPAJ, celle-ci n'est pas constante et d'authentiques dyschromatopsies de type rouge-vert ont été rapportées dans cette pathologie. L'étude du champ visuel met en évidence un scotome central dont l'étendue et la profondeur dépendent de l'acuité visuelle résiduelle. Enfin, le bilan électrophysiologique confirme le dysfonctionnement des CRG avec une altération spécifique de l'électrorétinogramme pattern et des réponses réduites ou absentes des PEV standard.
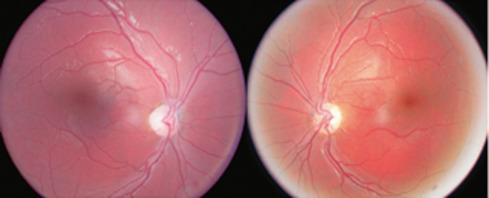
Fig. 21-29 Fond d’oeil d’une atrophie optique.
Rétinophotographie d’une patiente de 6 ans porteuse d’une AOD par mutation du gène OPA1. Il existe une pâleur papillaire diffuse plus marquée en temporal.
L'AOD/OPAJ est le plus souvent isolée. Cependant, dans près de 20 % des cas, des troubles associées peuvent être retrouvés. Il s'agit majoritairement de troubles de l'audition observés chez plus de 6 % des patients AOD/OPAJ [5]. Ces troubles de l'audition peuvent être infracliniques, d'âge de survenue variable, ou congénitaux et sévères avec une variabilité de présentation au sein d'une même fratrie. Une mutation faux sens du gène OPA1 est fréquemment retrouvée et notamment la mutation R445H (c.1334G>A) présente dans près de la moitié des observations [5]. Les potentiels évoqués auditifs (PEA) sont altérés témoignant d'une atteinte des voies auditives alors que les auto-émissions restent préservées confirmant la préservation des cellules ciliées de l'oreille interne. La population pédiatrique est peu concernée par l'AOD plus : association d'une AOD/OPAJ typique dans son âge d'apparition et sa présentation à des troubles neurologiques, ces derniers étant souvent d'apparition retardée vers la troisième décennie. Au maximum, ces troubles neurologiques en imposent pour une sclérose en plaques ou réalisent une paraplégie. Mais ils sont souvent limités à la présence d'une neuropathie sensitive ou motrice, d'une myopathie proximale, de troubles cérébelleux avec ataxie ou d'une ophtalmoplégie externe [7]. Des troubles cardiaques à type de syndrome de préexcitation ont également été signalés.
Quelle que soit la forme clinique de l'AOD/OPAJ, la BAV semble irréversible, bien que quelques rares cas de récupération aient été rapportés [8]. Il n'existe à ce jour aucun traitement disponible, même si l'intérêt de l'idébénone a été signalé.
La BAV apparaît généralement avant l'âge de 10 ans et présente une évolution lente quoi qu'un peu plus rapide que celle de l'AOD/OPA1. L'acuité visuelle peut atteindre 1/10 avant l'âge de 40 ans. Cette BAV est due à l'atrophie optique ainsi qu'à la présence constante d'une cataracte, habituellement diagnostiquée avant l'âge de 10 ans. Son retentissement sur la fonction visuelle est extrêmement variable puisqu'elle peut être poussiéreuse, corticale antérieure et postérieure, sous-capsulaire antérieure et postérieure, céruléenne, etc. La date du geste chirurgical varie de l'enfance à plus de 50 ans. La papille optique est atrophique avec une pâleur parfois limitée à son secteur temporal, ce que confirment les rares études en OCT. L'examen du champ visuel retrouve un scotome central plus ou moins profond et l'étude du sens chromatique met en évidence une dyschromatopsie acquise sans axe défini. Des troubles neurologiques, extrapyramidaux, à type de spasticité ou d'ataxie, ont également été rapportés mais ils apparaissent généralement chez des patients adultes. Il en est de même des troubles auditifs observés chez près de 20 % des patients dans une série anglaise [12].
L'évolution clinique des AOD dues à des mutations de ces gènes semble assez similaire à celle observée au cours de l'AOD/OPAJ. La BAV apparaît habituellement entre la première et la troisième décennie et présente par la suite une évolution progressive. À la phase d'état de la maladie, les papilles sont atrophiques ou pâles dans le secteur temporal. L'étude du sens chromatique retrouve inconstamment une dyschromatopsie de type « bleue-jaune » . Les réponses des PEV s'altèrent progressivement avec l'évolution de la pathologie. Un déficit auditif d'apparition tardive est volontiers retrouvé chez les patients porteurs d'une mutation du gène OPA8.
Avec une répartition ubiquitaire et une prévalence estimée à 1 cas pour 50 000 habitants, la neuropathie optique de Leber (NOHL) est sans doute la seconde forme de NOH isolée par ordre de fréquence. Elle affecte plus volontiers les hommes que les femmes, sans que cette prédominance masculine ne soit encore expliquée. La présence d'haplotypes de gènes protecteurs du génome mitochondrial localisés sur le chromosome X ou le rôle des œstrogènes ont été évoqués mais non confirmés [13]. Cette différence de sex-ratio varierait d'un pays à l'autre, étant de 8:2 en Europe et aux États-Unis et de 6:4 au Japon.
La transmission de la NOHL est secondaire à la présence de mutations ponctuelles au niveau de l'ADNmt. Mais, d'autres facteurs participeraient à son développement : facteurs génétiques (interactions avec des gènes contrôlant le génome mitochondrial), anatomiques ou environnementaux/toxiques [1].
Les mutations de l'ADNmt, recherchées sur un simple prélèvement sanguin, intéressent principalement les gènes des différentes sous-unités de la NADH (nicotinamide adenine dinucleotide hydrogen) déshydrogénase ou complexe I de la chaîne respiratoire. Il existe des mutations dites « primaires » ou « causales » dont la présence est nécessaire, mais non suffisante, au développement d'une NOHL. En effet, toute personne porteuse d'une mutation primaire ne développe pas une NOHL. Néanmoins, il est parfois retrouvé chez des apparentés sains porteurs de mutations de l'ADNmt des télangiectasies péripapillaires et un œdème de la couche des fibres optiques en OCT [14]. Leur présence ne préjuge pas d'un risque accru de déclencher une NOHL. Ces anomalies peuvent correspondre à un mécanisme de compensation visant à lutter contre le dysfonctionnement mitochondrial ou à une phase de stase axonale pouvant précéder une éventuelle décompensation et l'apparition d'une BAV.
Trois mutations, ND4/G11778A, ND1/G3460A et ND6/T14484C, sont responsables d'environ 95 % des NOHL. D'autres mutations, plus rares, peuvent être recherchées en cas de normalité du bilan initial. Mais, certaines sont encore méconnues. La normalité du bilan génétique ne permet donc pas d'éliminer le diagnostic de NOHL.
Le nombre de copies d'ADNmt mutées au sein d'une cellule, appelé taux d'hétéroplasmie (ou homoplasmie quand toutes les copies sont mutées), semble influer sur le risque de développer une NOHL. Ce risque serait plus important en cas de seuil d'hétéroplasmie supérieur à 75 % . Mais ce taux d'hétéroplasmie varie d'un tissu à l'autre et d'une cellule à l'autre. La valeur retrouvée lors d'un prélèvement sanguin n'est pas un bon reflet de ce qui se passe dans les autres tissus de l'organisme, notamment au sein du nerf optique. Le taux d'hétéroplasmie peut varier de 33 % dans le sang à 95 % dans le nerf optique.
Il est admis que la mutation primaire retrouvée peut modifier le tableau clinique de la NOHL. La fréquence des récupérations serait plus faible en présence d'une mutation 11778 et plus élevée avec les mutations 3460 ou 14484. Cependant, il existe de grandes variations de l'évolution de la fonction visuelle au sein d'une même fratrie porteuse de la même mutation. Le risque de développer une NOHL induite par les toxiques, notamment par l'alcool ou le tabac dont le rôle reste controversé, serait accru avec les mutations 3460 et 14484 [15]. Néanmoins, les prises aiguës et importantes d'alcool (bindge drinking) favoriseraient les BAV.
Des mutations, dont l'existence n'est pas suffisante pour qu'apparaisse une NOHL, sont dites « secondaires » et se comportent comme des « polymorphismes modificateurs » , modulant la réponse à une mutation primaire. Les haplogroupes et sous-haplogroupes de l'ADNmt ont un rôle identique et influent sur l'évolution de la pathologie.
Seules les femmes transmettent les mutations de l'ADNmt, puisque toutes les mitochondries de l'organisme proviennent de l'ovule et sont transmises à tous les enfants.
De nombreux cas de NOHL apparaissent sporadiques et des antécédents familiaux ne sont retrouvés que chez 37 à 89 % des patients. Ce pourcentage dépendrait du taux d'hétéroplasmie de l'ADNmt au sein de l'ovule. Il est admis que le risque d'avoir un enfant atteint de NOHL est faible lorsque ce taux est faible et/ou lorsqu'une femme présente moins de 80 % de son ADNmt muté. Au sein de la fratrie d'un patient atteint, le risque de développer une NOHL varie de 30 à 83 % pour un homme et de 5 à 32 % pour une femme.
Le conseil génétique est donc difficile face à cette pathologie. Il est néanmoins possible d'affirmer, du fait du mode de transmission « non mendéléienne » qu'un homme atteint n'a aucun risque pour sa descendance.
La présentation clinique de la NOHL est assez stéréotypée. Mais, il existe de nombreuses formes atypiques, notamment chez l'enfant, qui peuvent égarer le diagnostic en l'absence d'antécédents familiaux.
La NOHL débute entre 15 et 30 ans. Mais des formes à début tardif, après 60 ans, ont été décrites ainsi que des formes pédiatriques apparaissant avant l'âge de 1 an [16]. La BAV apparaît brutalement, en quelques jours, d'emblée profonde, comprise entre 1/50 et la perception lumineuse. Celle-ci intéresse les deux yeux en même temps chez près de 50 % des patients. Lorsqu'elle est initialement unilatérale, l'œil adelphe est touché après un délai moyen de 2 mois. Les formes strictement unilatérales sont exceptionnelles, hormis chez l'enfant.
En effet, chez les enfants les plus jeunes, la NOHL peut être atypique, d'évolution insidieuse, avec une BAV volontiers progressive sur plusieurs semaines et cliniquement apparemment unilatérale. Il n'est pas rare de devoir l'évoquer devant un enfant ayant une BAV asymétrique et globalement peu sévère, dont il est impossible de préciser le mode évolutif ou la date éventuelle d'apparition. Devant une telle situation, il faut éliminer les autres étiologies d'atteinte du nerf optique. À l'inverse, la BAV peut être d'emblée totale. Ces présentations spécifiques seraient dues à des particularités anatomiques des papilles de l'enfant. Les formes aiguës seraient associées à des papilles dont la surface et le diamètre vertical seraient plus petits que ceux de sujets témoins ou d'enfants présentant une forme progressive.
Au stade initial, l'examen du fond d'œil peut retrouver une papille normale ou ayant un aspect « pseudo-œdémateux » (fig. 21-30). Mais un examen attentif révèle que cet aspect est dÛ à la présence de télangiectasies péripapillaires (fig. 21-31). Si elle peut être réalisée, l'angiographie à la fluorescéine confirme l'absence de fuite de colorant. Ces télangiectasies pourraient traduire un stade préclinique de la maladie et leur présence au niveau de l'œil apparemment sain, dans les formes « unilatérales » , constitue un argument diagnostic important. Elles sont également observées chez des apparentés sains porteurs d'une mutation primaire de l'ADNmt. Ces anomalies microvasculaires papillaires disparaissent quand s'installe l'atrophie optique (AO) isolée sans excavation papillaire qui représente l'évolution naturelle de la papille lors de la NOHL (fig. 21-31). Cette AO intéresse la totalité du disque ou reste limitée à son secteur temporal. Une telle présentation est volontiers retrouvée chez les patients présentant une amélioration secondaire de l'acuité visuelle mais n'a pas de valeur pronostic. La survenue d'une maculopathie évoquant une maladie de Stargardt, a été rapportée en présence des mutations 11778 ou 15257 de l'ADNmt.
Les examens complémentaires confirment l'atteinte du nerf optique et permettent d'éliminer une autre étiologie. L'OCT retrouve au stade initial de la maladie un pseudo-œdème de la couche des fibres optiques, également retrouvé chez les patients apparentés sains (fig. 21-32) [14]. Après 6 mois d'évolution, alors qu'existe une atrophie optique, l'épaisseur de la couche des fibres optiques diminue, principalement dans le secteur interpapillomaculaire. L'étude du sens chromatique retrouve une dyschromatopsie rouge-vert de type 2. Un vaste scotome central absolu et relatif d'étendue variable peut être détecté lors du relevé du champ visuel manuel ou automatisé (fig. 21-33). Aucune réponse discernable ne peut être enregistrée aux PEV alors que l'électrorétinogramme (ERG) est normal ou retrouve une altération de type photopique y compris en l'absence d'anomalie maculaire clinique. L'IRM cérébrale doit être systématiquement réalisée. Elle peut être normale, confirmer l'atrophie des nerfs optiques après plusieurs mois d'évolution de la NOHL ou objectiver la présence d'hypersignaux de la substance blanche en T2 au niveau de tout l'encéphale, du chiasma et des nerfs optiques. Leur présence pose des problèmes diagnostiques avec la sclérose en plaques, en particulier lors de formes particulières dites « Leber plus » .
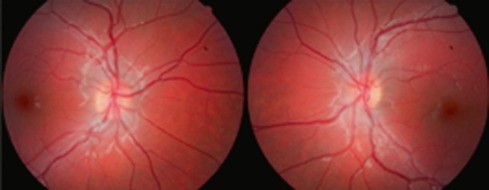
Fig. 21-30 Fond d’oeil en présence d’une neuropathie optique héréditaire de leber.
Rétinophotographie d’un patient de 9 ans porteur de la mutation 11778 de la NOHL. Aspect de faux oedème papillaire.
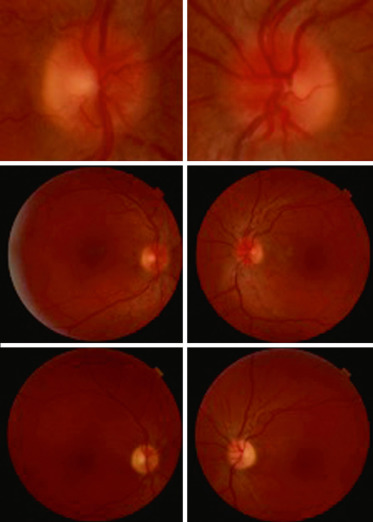
Fig. 21-31 Fond d’oeil en présence d’une neuropathie optique héréditaire de leber.
Rétinophotographie d’un patient porteur de la mutation 11778 de la NOHL, présentant une baisse d’acuité visuelle droite depuis 1 mois, gauche depuis 15 jours. En haut : noter l’aspect hyperhémié de la papille secondaire aux télangiectasies, réalisant un pseudo-oedème. Le patient présente déjà un début de pâleur temporale droite. Au milieu et en bas, rétinophotographies initiales et un an plus tard : noter la disparition de l’hyperhémie papillaire aux dépens d’une atrophie globale, non creusante, prédominant en temporal. Noter également la perte de réflectivité des fibres ganglionnaires partant du quadrant temporal des nerfs optiques.
(Coll. P. Lebranchu.)
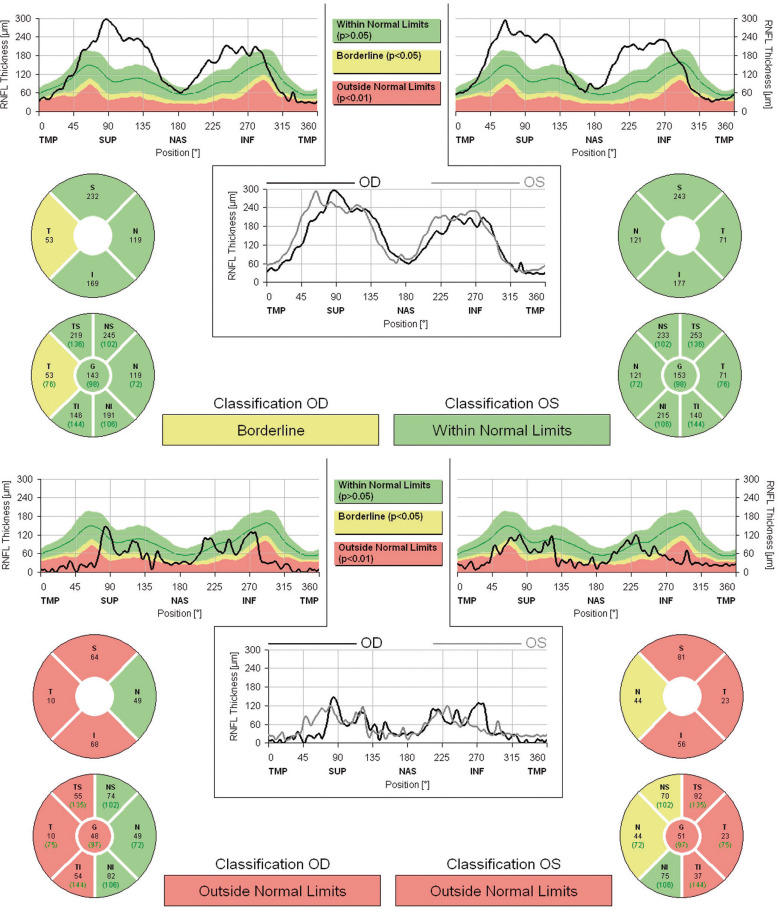
Fig. 21-32 OCT du nerf optique en présence d’une neuropathie optique héréditaire de Leber.
OCT RNL du même patient. En haut : OCT le jour de la 1re consultation, confirmant l’aspect de pseudo-oedème du nerf optique. Noter le début d’atrophie temporale apparue entre le moment de la baisse de vision (1 mois pour l’oeil droit, 15 jours pour l’oeil gauche) et le moment où le patient est adressé pour prise en charge. En bas : OCT un an plus tard. Noter l’atrophie globale du nerf optique, prédominant dans le quadrant temporal.
(Coll. P. Lebranchu.)
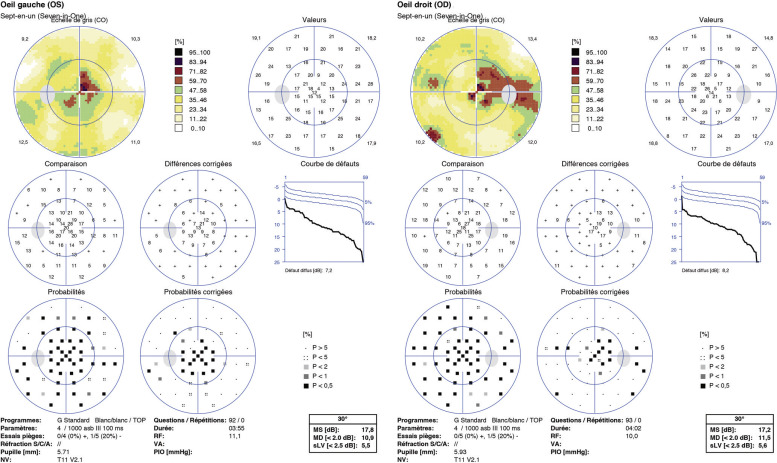
Fig. 21-33 Champ visuel automatisé en présence d’une neuropathie optique héréditaire de Leber.
Champs visuel droit et gauche du même patient (Octopus®, stratégie Top). Le patient présente de façon bilatérale un déficit diffus, un scotome cæco-central et un abaissement du seuil fovéolaire.
(Coll. P. Lebranchu.)
Habituellement, la BAV est permanente lors de la NOHL. Il est volontiers décrit des fluctuations plus ou moins marquées de l'acuité visuelle et de la taille du scotome central évoquant un pseudo-syndrome de Uhthoff. Néanmoins, des récupérations unilatérales ou asymétriques et plus ou moins importantes de la fonction visuelle ont été rapportées, parfois plusieurs mois ou années après l'installation de la maladie. Elles sont exceptionnellement « totales » . La fréquence de ces récupérations est mal connue et varierait selon la mutation primaire. Elle serait plus élevée dans les formes d'apparition précoce et chez les femmes [17]. Mais, elles ne sont nullement systématiques ni totales dans les formes pédiatriques. Enfin, il existe des différences au sein d'une même fratrie portant la même mutation primaire. Elles s'accompagnent d'une fenestration du scotome central qui est d'autant plus importante que l'acuité visuelle finale est meilleure.
Habituellement isolée, la NOHL peut parfois s'accompagner de manifestations extra-oculaires, voisines de celles observées lors de l'AOD/OPA1, qui doivent être recherchées, y compris chez l'enfant même si leur fréquence est mal connue dans cette population. Il s'agit d'une part de troubles du rythme cardiaque à type de syndrome de préexcitation (syndromes de Wolf-Parkinson-White ou de Lown-Ganong-Levine). Par ailleurs, il est rapporté des anomalies neurologiques, parfois minimes asymptomatiques à type de calcification des noyaux gris, de surdité, d'ataxie, d'atteinte musculaire, purement histologique ou plus rarement clinique, parfois plus importantes sous forme de dystonie ou de manifestations pouvant en imposer pour une SEP dans le cadre du « Leber plus » . Il peut être difficile de différencier ces deux affections responsables d'atteinte des nerfs optiques et d'hypersignaux de la substance blanche en IRM cérébrale. Les relations entre ces deux pathologies restent très controversées [18].
Il n'existe aucun traitement reconnu de la NOHL. Différentes molécules (vitamine B2, vitamine B12 ou co-enzyme Q10) ont été testées sans démontrer d'efficacité. Celle de l'idébénone reste discutée [19]. Un dérivé du co-enzyme Q10, l'EPI-743, semble prometteur [20]. La ciclosporine, en cours d'évaluation, donnerait des résultats encourageants pour limiter l'atteinte de l'œil adelphe dans les formes à début différé [6]. La thérapie génique constitue une autre voie thérapeutique en cours d'évaluation [21]. La prévention joue donc un rôle important. Elle vise à supprimer toute situation augmentant la consommation d'énergie par la cellule et, en premier lieu, les toxiques, même si le rôle de l'alcool et du tabac est secondaire dans la population pédiatrique. Le rôle du stress reste controversé car difficilement quantifiable.
Seules trois fratries porteuses d'une NOH liée au chromosome X (OPA2) ont été rapportées dans la littérature [4]. Tous les hommes atteints sont porteurs d'une AO et d'une BAV progressive d'apparition précoce, dès la première année de vie et parfois dès la naissance. Des troubles neurologiques très discrets (anomalie des réflexes ostéotendieux, syndrome cérébelleux) et un retard mental sont également signalés dans une fratrie. Toutes les femmes conductrices sont cliniquement asymptomatiques. Le gène responsable est situé en Xp11.4-p11.21.
À ce jour, dans le cas de l'atrophie optique récessive (AOR), quatre gènes ont été localisés et certains ont été identifiés. L'un de ces gènes, RTN4IP1 donne une forme classique d'apparition très précoce. Les autres sont responsables d'une symptomatologie d'évolution plus ou moins rapide débutant généralement au cours de la première décennie. Trois d'entre eux, ACO2, WFS1 et SCA7 sont également responsables de NOH symptomatiques.
Cette forme, qui n'est peut être pas la plus fréquente, correspond à la description classique des AOR. Ses premières manifestations apparaissent précocement au cours de la première année de vie et sont parfois congénitales. C'est pourquoi elle doit être évoquée devant un enfant présentant un comportement de malvoyant : indifférence à son entourage, absence de sourire réponse ou de poursuite des objets. La présence d'un nystagmus de malvoyant confirme que l'acuité visuelle est effondrée.
L'examen du fond d'œil retrouve une papille de taille normale, non excavée mais totalement atrophique (éliminant une hypoplasie papillaire) (fig. 21-34). La présence d'une consanguinité ne constitue pas un élément diagnostique à ce stade puisqu'elle s'observe lors de nombreuses autres étiologies de malvoyance de l'enfant, dont le nystagmus congénital (mais au cours de celui-ci, le bilan électrophysiologique est normal). Or, les PEV sont non discernables du bruit de fond alors que l'ERG est normal, éliminant une malvoyance du jeune enfant à fond d'œil sensiblement normal (amaurose congénitale de Leber ou achromatopsie notamment). Le bilan neuroradiologique est sans particularité. La fonction visuelle reste stable ou ne présente qu'une lente dégradation. Les papilles optiques peuvent s'excaver avec l'âge. Cette AOR reste habituellement isolée.
Une mutation du gène RTN4IP1 a été retrouvée dans une forme de survenue précoce au sein de neuf fratries principalement d'origine tzigane présentant un tableau proche de cette forme classique. La BAV est précoce, parfois congénitale [22].
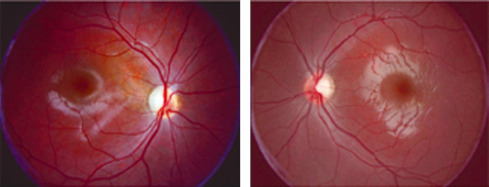
Fig. 21-34 Fond d’oeil d’une atrophie optique récessive.
Rétinophotographie d’un garçon de 11 ans retrouvant une atrophie papillaire. Gène causal inconnu.
Le gène d'AOR, OPA6 localisé sur le chromosome 8 en 8q21-q22, est responsable d'une forme isolée de NO débutant au cours de la première décennie [23]. La BAV est initialement peu importante, mais l'acuité visuelle se dégrade progressivement au cours de la vie pour aboutir à un handicap visuel franc aux alentours de 50 ans. Les papilles optiques évoluent rapidement vers l'atrophie avec une atteinte prédominante du faisceau interpapillomaculaire. Le champ visuel confirme la présence d'un scotome central.
Le gène d'AOR, OPA7 encore dénommé TMEM126A est localisé en 11q14.1-q21 [24]. Il s'exprime dans la mitochondrie et code une protéine dont la fonction est inconnue. Ses mutations sont responsables d'une AOR débutant entre 2 et 6 ans et d'évolution rapide et sévère (avec une acuité visuelle comprise entre 1/10 et compte les doigts). Les papilles optiques sont atrophiques. Le champ visuel retrouve un scotome central qui augmente lentement avec perte de vision. Des anomalies cardiaques (cardiomyopathie, syndrome de Wolff-Parkinson-White), auditives (surdité modérée) ou neurologiques/neuroradiologiques (hypersignaux de la substance blanche) ont été rapportées.
L'AOR OPA9 due à des mutations du gène ACO2 a été décrite chez deux patients [25]. Elle débute entre 3 et 5 ans et entraîne une altération isolée de la fonction visuelle, stable après 20 ans. L'acuité visuelle est proche de 1/10 avec présence d'un scotome paracentral. Il est noté une dyschromatopsie de type rouge-vert. Les papilles sont atrophiques avec une perte de fibres prédominant en temporal.
Il a été rapporté la survenue d'AOR strictement isolées associées à des mutations du gène WFS1 (responsable par ailleurs du syndrome de Wolfram [SW]) ou du gène SCA7 responsable de l'ataxie spinocérébelleuse (ASC) de type 7.
Certaines NOH syndromiques sont dues à la présence de mutations dans des gènes spécifiques. Mais deux sont associées à la présence de mutations dans des gènes également responsables de NOH isolées.
Le gène WFS1 code la wolframine, protéine localisée au niveau de la membrane du réticulum endoplasmique (RE) où il régule l'entrée du calcium. Mais des interactions entre RE et mitochondries ont pu être retrouvées. Le gène CISD2, ou WFS2,, code une protéine s'exprimant également un niveau du RE mais dont l'action reste mal connue.
Initialement appelé DIDMOAD (diabetes insipidus, diabetes mellitus, optic atrophy, and deafness), le SW est transmis selon un mode récessif autosomique. Son diagnostic est porté devant la présence de deux critères majeurs ou d'un critère majeur et de deux critères mineurs. Les critères majeurs incluent : un diabète insulino-dépendant peu sévère retrouvé entre 3 et 16 ans et une atrophie optique apparaissant entre 10 et 16 ans (fig. 21-35) [26, 27]. Mais elle peut apparaître au-delà de 20 ans. A contrario, l'atteinte du nerf optique peut apparaître dès la première année de vie. Les critères mineurs associent une surdité minime à modérée et un diabète insipide parfois infraclinique, apparaissant tous deux durant la seconde décennie et des troubles urinaires, insidieux avant 20 ans et devenant gênant vers 25-30 ans [26,28 ]. Ils sont aggravés par les troubles neurologiques, plus tardifs, également responsables d'une ataxie, de neuropathies périphériques, d'un déficit cognitif et peuvent mettre en jeu le pronostic vital du fait des troubles ventilatoires [26]. L'existence de troubles psychiatriques chez les patients ou les apparentés est évoquée. La mise en évidence de deux mutations, parfois différentes (double hétérozygote) du gène et notamment d'au moins une mutation aboutissant à une perte de fonction de la protéine, confirme le diagnostic.
L'évolution de la neuropathie optique est généralement assez lente. L'acuité visuelle basse au départ devient progressivement inférieure à 1/20, alors que le scotome central se majore en profondeur et surface. Il a également été rapporté l'existence de cataractes précoces.
Il n'existe aucun traitement permettant de limiter l'évolution de la neuropathie optique. Une thérapie génique est actuellement à l'étude.
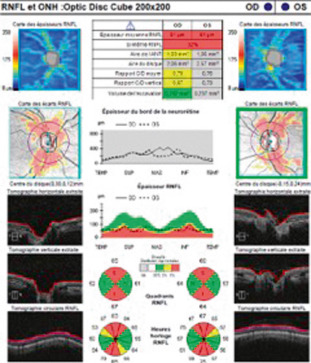
Fig. 21-35 OCT du nerf optique en présence d’un syndrome de Wolfram.
Il existe une diminution des fibres optiques chez un adolescent.
Sous ce terme, sont regroupés des SW récessifs autosomiques dont la présentation est incomplète, moins sévère et d'apparition plus tardive et des SW dominants autosomiques également de présentation incomplète, moins sévère et d'apparition plus tardive. Le diagnostic est porté devant la présence d'un seul critère majeur parmi ceux présentés précédemment et d'un critère mineur. Le pronostic visuel et extra-oculaire de ces patients n'est pas totalement connu.
Cette forme autosomique récessive plus rare que celle due à une mutation du gène WFS1 est principalement observée dans les populations d'origine libanaise et jordanienne. Les principaux critères majeurs et mineurs sont identiques en dehors de l'absence de diabète insipide. Il est également noté des ulcères gastriques responsables d'hémorragies digestives.
Cette pathologie rare, autosomique récessive, principalement retrouvée dans la population juive iraquienne est due à la présence de mutations particulières dans le gène OPA3. Il se caractérise par l'association d'une atrophie optique survenant au cours de la première décennie, lentement évolutive, à des troubles neurologiques (mouvements choréo-athétosique ou syndrome extrapyramidal), apparaissant également avant 10 ans. Mais il peut exister des formes à début plus tardif. L'évolution des troubles neurologiques aboutit à la survenue d'une paraplégie spastique, d'une ataxie et d'un déficit cognitif modéré. L'existence d'une acidurie 3-méthylglutaconique est caractéristique de ce syndrome et permet le diagnostic. Il n'existe aucun traitement spécifique.
Cette AOR « syndromique » est proche du syndrome de Costeff dans sa présentation clinique quoi qu'elle s'en différencie par quelques éléments. L'atrophie optique apparaît très précocement comme en témoigne l'existence d'un nystagmus de malvoyant. Elle reste d'intensité variable, de modérée à sévère et peu évolutive. Les troubles neurologiques apparaissent durant la seconde décennie et présentent des variations inter- et intrafamiliales. Ils associent syndrome pyramidal avec spasticité puis paraplégie, troubles vésicosphinctériens, syndrome extrapyramidal, ataxie, troubles sensitifs profonds, pieds creux, neuropathie périphérique et retard mental [22]. Leur évolution est progressive mais aboutit dans deux tiers des cas à une perte de déambulation vers l'âge de 25 ans et nécessite parfois des interventions spécifiques pour allonger le tendon d'Achille et faciliter la déambulation et la statique. L'IRM peut être normale ou révéler une atrophie cérébelleuse [22].
De nombreuses pathologies neurologiques pouvant débuter dans l'enfance s'accompagnent d'atrophie optique. Il faut citer l'ataxie de Friedreich, due à une mutation de la frataxine, débutant souvent vers 20 ans, mais dont le début peut être plus précoce avec une AO apparaissant à l'adolescence [29]. De même, différentes classes d'ASC, telles que l'ASC1 ou l'ASC7 liée à des mutations du gène SCA7 sont associées à des NOH syndromiques [30]. Quoique ces pathologies débutent généralement à l'âge adulte, des cas ont été rapportés en fin de l'adolescence. Les mutations du gène AFG3L2 responsables de l'ASC28, seraient également responsables de tableaux neurologiques associés à une AOD.
La neuropathie sensitive et motrice héréditaire VI, dominante autosomique, est une forme de maladie de Charcot-Marie-Tooth due à des mutations du gène NFM2 au cours de laquelle l'AO peut apparaître dans l'enfance sur un mode insidieux et progresser lentement. En revanche, lorsqu'elle débute à l'âge adulte, son évolution est plus rapide [25]. Nous ne ferons que citer la paraplégie spastique 7 secondaire à des mutations dans le gène SPG7 codant la paraplégine, mais qui est habituellement une pathologie de l'adulte (Tableau 21-9).
[1] Milea D, Amati-Bonneau P, Reynier P, Bonneau D Genetically determined optic neuropathies Curr Opin Neurol: ( 2010 ) :23: 24-28
[2] Ferre M, Bonneau D, Milea D Molecular screening of 980 cases of suspected hereditary optic neuropathy with a report on 77 novel OPA1 mutations Human Mutation: ( 2009 ) :30: E692-E705
[3] Yu-Wai-Man P, Griffiths PG, Burke A The prevalence and natural history of dominant optic atrophy due to OPA1 mutations Ophthalmology: ( 2010 ) :117: 1538-1546 1546.e1
[4] Votruba M Molecular genetic basis of primary inherited optic neuropathies Eye (Lond): ( 2004 ) :18: 1126-1132
[5] Leruez S, Milea D, Defoort-Dhellemmes S Sensorineural hearing loss in OPA1-linked disorders Brain: ( 2013 ) :136: e236-
[6] Meunier I, Lenaers G, Hamel C, Defoort-Dhellemmes S Les neuropathies optiques héréditaires : du signe clinique au diagnostic J Fr Ophtalmol: ( 2013 ) :36: 886-900
[7] Yu-Wai-Man P, Shankar SP, Biousse V Genetic screening for OPA1 and OPA3 mutations in patients with suspected inherited optic neuropathies Ophthalmology: ( 2011 ) :118: 558-563
[8] Lenaers G, Hamel C, Delettre C Dominant optic atrophy Orphanet J Rare Dis: ( 2012 ) :7: 46-
[9] Barboni P, Carbonelli M, Savini G OPA1 mutations associated with dominant optic atrophy influence optic nerve head size Ophthalmology: ( 2010 ) :117: 1547-1553
[10] Yu-Wai-Man P, Stewart JD, Hudson G OPA1 increases the risk of normal but not high tension glaucoma J Med Genet: ( 2010 ) :47: 120-125
[11] Toomes C, Marchbank NJ, Mackey DA Spectrum, frequency and penetrance of OPA1 mutations in dominant optic atrophy Human Molecular Genetics: ( 2001 ) :10: 1369-1378
[12] Sergouniotis PI, Perveen R, Thiselton DL Clinical and molecular genetic findings in autosomal dominant OPA3-related optic neuropathy Neurogenetics: ( 2015 ) :16: 69-75
[13] Shankar SP, Fingert JH, Carelli V Evidence for a novel X-linked modifier locus for leber hereditary optic neuropathy Ophthalmic Genet: ( 2008 ) :29: 17-24
[14] Barboni P, Savini G, Feuer WJ Retinal nerve fiber layer thickness variability in Leber hereditary optic neuropathy carriers Eur J Ophthalmol: ( 2012 ) :22: 985-991
[15] Kerrison JB, Miller NR, Hsu F A case-control study of tobacco and alcohol consumption in Leber hereditary optic neuropathy Am J Ophthalmol: ( 2000 ) :130: 803-812
[16] Reynier P, Penisson-Besnier I, Moreau C mtDNA haplogroup J : a contributing factor of optic neuritis Eur J Hum Genet: ( 1999 ) :7: 404-406
[17] Mashima Y, Hiida Y, Oguchi Y Lack of differences among mitochondrial DNA in family members with Leber's hereditary optic neuropathy and differing visual outcomes J Neuroophthalmol: ( 1995 ) :15: 15-19
[18] Vanopdenbosch L, Dubois B, D'Hooghe MB Mitochondrial mutations of Leber's hereditary optic neuropathy : a risk factor for multiple sclerosis J Neurol: ( 2000 ) :247: 535-543
[19] Klopstock T, Yu-Wai-Man P, Dimitriadis K A randomized placebo-controlled trial of idebenone in Leber's hereditary optic neuropathy Brain: ( 2011 ) :134: 2677-2686
[20] Sadun AA, Chicani CF, Ross-Cisneros FN Effect of EPI-743 on the clinical course of the mitochondrial disease Leber hereditary optic neuropathy Arch Neurol: ( 2012 ) :69: 331-338
[21] Ellouze S, Augustin S, Bouaita A Optimized allotopic expression of the human mitochondrial ND4 prevents blindness in a rat model of mitochondrial dysfunction Am J Hum Genet: ( 2008 ) :83: 373-387
[22] Copeliovitch L, Katz K, Arbel N Musculoskeletal deformities in Behr syndrome J Pediatr Orthop: ( 2001 ) :21: 512-514
[23] Barbet F, Gerber S, Hakiki S A first locus for isolated autosomal recessive optic atrophy (ROA1) maps to chromosome 8q Eur J Hum Genet: ( 2003 ) :11: 966-971
[24] Hanein S, Perrault I, Roche O TMEM126A, encoding a mitochondrial protein, is mutated in autosomal-recessive nonsyndromic optic atrophy Am J Hum Genet: ( 2009 ) :84: 493-498
[25] Rouzier C, Bannwarth S, Chaussenot A The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy ‘plus' phenotype Brain: ( 2012 ) :135: 23-34
[26] Barrett TG Mitochondrial diabetes, DIDMOAD and other inherited diabetes syndromes Best Pract Res Clin Endocrinol Metab: ( 2001 ) :15: 325-343
[27] Marshall BA, Permutt MA, Paciorkowski AR Phenotypic characteristics of early Wolfram syndrome Orphanet J Rare Dis: ( 2013 ) :8: 64-
[28] Ribière C, Kaboré FA, Chaussenot A Troubles vésicosphinctériens au cours du syndrome de Wolfram Prog Urol: ( 2013 ) :23: 519-523
[29] Fortuna F, Barboni P, Liguori R Visual system involvement in patients with Friedreich's ataxia Brain: ( 2009 ) :132: 116-123
[30] Klebe S, Depienne C, Gerber S Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy Brain: ( 2012 ) :135: 2980-2993
Tableau 21-9 – Âge d’apparition, mode évolutif des neuropathies optiques héréditaires de l’enfant et manifestations extra-oculaires associées
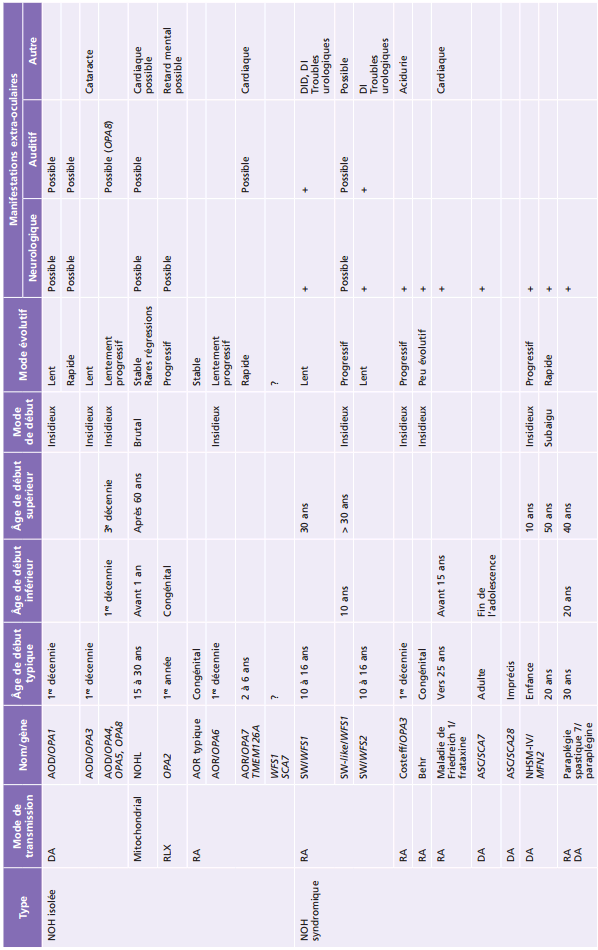
AOD : atrophie optique dominante ; AOR : atrophie optique récessive ; ASC : ataxie spinocérébelleuse ; DA : dominant autosomique ; DI : diabète insipide ; DID : diabète insulino-dépendant ; NOH : neuropathies optiques héréditaires ; NOHL : neuropathie optique héréditaire de Leber ; RA : récessif autosomique ; RLX : récessif lié au chromosome X ; SW : syndrome de Wolfram.